Úvod
Glykogen je polymer glukózy, který se nachází zejména v játrech (10 % hmotnosti, zásoby vydrží cca 18 hodin hladovění a spotřebuje se až k nule) a svalech (1 % hmotnosti, působí zde jako zdroj energie a nikdy se nespotřebuje zcela). Glukóza je v glykogenu vázána formou 1,4 vazeb a po cca 10 glukózových zbytcích dochází k větvení formou 1,6 vazby. Glykogen je skladován v 10 – 40 nm granulích, které obsahují i enzymy a strukturální protein. Po glykogenolýze je cca 10 % glykogenu v granulu zachováno jako matrix pro budoucí glykogenogenezi.

Glykogenolýza
- 1a. Z molekuly glykogenu se od konce s glukózou s volnou OH skupinou odštěpují pomocí enzymu glykogenfosforylázy (štěpí 1,4 vazby, fosforyluje) jednotlivé molekuly glukózy spolu jejich fosforylací za vzniku glukóza-1-fosfátu (G-1-P).
- 1b. Jakmile na štěpeném řetězci zbývají poslední čtyři glukózy, jsou poslední tři odštěpeny un-block a přesunuty na sousední volný OH konec glukózy jiného řetězce pomocí enzymu α1,6-transglykosylázy (linearizační enzym). Stejný enzym nakonec odštěpí glukózu na poslední 1,6 vazbě (bez fosforylace) a původní řetězec zaniká.
- 2. G-1-P je následně fosfoglukomutázou převeden na glukóza-6-fosfát (G-6-P).
- 3. G-6-P je buď enzymem glukóza-6-fosfatáza přeměněn na glukózu (pouze v játrech nikoliv svalu a mozku), nebo vstupuje do anaerobní glykolýzy nebo pentosafosfátového cyklu.
Glykogenogeneze
Syntéza glykogenu začíná na malém polymeru glukózy (nejméně čtyřčlenný), který se nazývá primer a až ten se následně prodlužuje glykogensyntázou. G-1-P nemá dostatečnou energii k vazbě k molekule glykogenu, k tomu je potřeba energeticky ještě bohatší forma uridindifosfátglukóza (UDP~G), která vzniká pomocí enzymu UDP-glukosadilfosforyláza. UDP-G je následně pomocí glykogensyntázy vázána na primer 1-4 vazbou. Při vzniku větvení se účastní větvící enzym, který indukuje vznik 1-6 vazeb.
Regulace – štěpení stimuluje a syntézu tlumí glukagon, adrenalin, thyroxin, opačně působí inzulín.
Celková prevalence glykogenóz (GSD) je ~ 1 : 20000 porodů. Až na výjimky (viz. níže) jsou AR dědičné.
1. Jaterní glykogenózy
Typ IX
Způsoben deficitem fosforyláza kinázy (PhK, enzym glykogenolýzy, čtyři podjednotky α, β, γ, δ), která aktivuje glykogenfosforylázu štěpící 1,4 vazby glykogenu za vzniku G-1-P. Existuje celkem sedm typů:
- Nejčastější je X vázaná forma s defektní podjednotkou α, která se projeví v 1 – 5 letech dítěte
- hepatomegalií a růstovou retardací. Akumulovaný glykogen v játrech má při histologickém vyšetření spíše roztříštěný vzhled (na rozdíl od dalších jednotek). S postupujícím věkem dochází k postupné normalizaci hepatomegalie i laboratorního nálezu.
- Jelikož je postižena glykogenolýza, je výrazná tendence k hypoglykémiím s kompenzačním snížením hladiny inzulínu a zvýšením ketolátek při lačnění s mírnou elevací lipidů i jaterních enzymů. Laktát i kyselina močová bývají v normě (nevzniká nadbytek G-6-P). Prognóza je obecně dobrá, u některých pacientů může naopak jaterní postižení progredovat do cirhózy a jaterního selhání. Proto je vhodné, aby dospělí pacienti byli pravidelně monitorování CT nebo MRI jater. Terapie je symptomatická, nutné jsou časté porce jídla k prevenci hypoglykémie (glukózu a ketony je nutné hlídat během epizod stresu). Dle posledních studií je časné zahájení podávání kukuřičného škrobu a proteinů spojeno s menším množstvím pozdních komplikací.
- Dalšími formami jsou závažné AR formy jaterní, svalové, jaterně-svalové a kardiální (typ VI, letální průběh během dětství).
Typ III (Coriho choroba)
Etiopatogeneze – AR dědičná choroba způsobená deficitem delinearizačního enzymu (součást glykogenolýzy, odpovědný za štěpení glykogenu v místě 1,6 vazeb).
Klinický obraz – projevuje se hypoglykémií, hepatomegalií, nízkou postavou a variabilně myopatií a kardiomyopatií. Pokud jsou postiženy játra a svaly, označuje se jako IIIa, pokud dominuje pouze postižení jater, označuje se IIIb. Na rozdíl od typu I:
- nevzniká nadbytek G-6-P, proto bývá laktát v normě (proto je v normě i kyselina močová).
- běžná je ketoacidóza (utlumení sekrece inzulínu při hypoglykémii), AST a ALT je zvýšené.
- také bývají polycystická ovaria, ale je častější kompletní PCO syndrom (hirsuitismus, nepravidelná ovulace). I zde bývá fertilita zachována.
U většiny pacientů se hepatomegalie postupně upravuje, v pozdním věku se může objevit i cirhóza jater s karcinomem (jaterní adenomy jsou vzácnější než u typu I).
Diagnostika a terapie – typ IIIa může být prokázán biopsií jater, svalů a srdce, typ IIIb pouze jater. Již v časných fázích choroby lze nalézt hepatocyty napěchované glykogenem s okrsky fibrózy. Další možností je genetické vyšetření. Specifická farmakoterapie neexistuje, léčbou může být vysokoproteinová dieta k nastartování glukoneogeneze a léčba komplikací. Úloha transplantace jater zůstává sporná.
Existují i ostatní typy GSD (viz. níže).
Typ I (von Gierkeho choroba)
Etiopatogeneze + klinický obraz – AR dědičná porucha glykolýzy při deficitu glukóza-6-fosfatázy (pouze v játrech, proto nejsou svalové příznaky ani kardiomyopatie) s incidencí 1 : 60 – 100 tisíc porodů. Lze rozlišit podtyp Ia (defektní enzym) a Ib (defektní transport G6P přes membránu mikrozomu).
- Dochází k bloku vzniku glukózy na úrovní G6P. Nadbytečný glykogen se díky tomu hromadí zejména v játrech, ledvinách a sliznici střeva (výrazná hepatomegalie od dětství). Při nemožnosti hydrolýzy na glukózu vstupuje nadbytečný G6P do anaerobní glykolýzy se vznikem pyruvátu a laktátu a laktátové acidózy (výraznější při horečkách a po podání glukagonu). Nadbytečný laktát kompetuje v renálních tubulech s urátem o transportér, což vede k hyperurikémie s rizikem časné dny.
- Vzniká i hypoglykémie, na kterou se organismus adaptuje poklesem sekrece inzulínu. Díky tomu se aktivuje lipáza v tukové tkáni se vznikem dyslipidémie (zvýšené LDL,TAG, fosfolipidy i volné mastné kyseliny). Nedostatek inzulínu může vést i k rozvoji ketoacidózy.
- Děti mají podobu panenky s tučnými tvářemi, tenké končetiny, malou výšku a prominující bříško díky výrazné hepatomegalii, zpomaluje se růst a opožďuje puberta.
- Je přítomna tendence k modřinám a krvácení, hyperurikémie, dyslipidémie. Běžné je oddálení puberty a cysty ovárií u většiny žen, ovšem se zachováním fertility (vyjádřený syndrom PCO s akné a hirsuitismem je naopak vzácný). Je zvýšené riziko pankreatitidy (↑ TAG), kardiovaskulárních chorob (↑ LDL), osteopenie až osteoporózy, od 25 let vznikají jaterní adenomy, které se mohou i malignizovat, od 20 let dochází k poklesu renálních funkcí s proteinurií s rizikem selhání ledvin.
- Pacienti s typem Ib mají navíc neutropenii vedoucí k recidivujícím infekcím a ústním a střevním ulceracím s průjmy. Z dlouhodobého hlediska je častá dna.

Diagnostika a terapie – klinický obraz + zvýšená hladina laktátu + dyslipidémie. Definitivní průkaz dá genetické vyšetření a biopsie jater se stanovením aktivity G6P. Terapie je založena na symptomatické léčbě jednotlivých příznaků a komplikací, v jednotlivých případech byl zaznamenán úspěch transplantace jater.
2. Svalové glykogenózy
Typ V (McArdleho choroba)
Etiopatogeneze – při AR dědičném deficitu svalové fosforylázy (součást glykogenolýzy, štěpí 1,4 vazby glykogenu za vzniku G-1-P) dochází k hromadění svalového glykogenu jako jejího hlavního substrátu s následným deficitem energie díky nedostatečné tvorbě ATP.
Klinický obraz – první příznaky většinou vznikají v dospělosti a patří mezi ně intolerance intenzivní fyzické námahy s křeče svalů, naopak mírná a střední námaha bývá tolerována. Pacient typicky popisuje, že po krátkém odpočinku chytá “druhý dech“. U 1/3 pacientů je bolest trvalá a ruší i spánek, u jedné poloviny pacientů se po cvičení objevuje načervenalá moč, jako důsledek myoglobinurie (těžká myoglobinurie po fyzickém excesu může vést až k renálnímu selhání). Přestože myokard nebývá postižen, občas bývá u těchto pacientů popisována hypertrofická kardiomyopatie.
Diagnostika – v klidu je lehce zvýšená hladina CK, po fyzické aktivitě se zvyšuje CK, myoglobin, amoniak a kyselina močová (důsledek zvýšeného obratu nukleových kyselin), naopak laktát se nezvyšuje ani po velkém výkonu. Diagnózu lze potvrdit průkazem enzymatického defektu při svalové biopsii nebo genetickým vyšetřením.
Defekt metabolismu glykogenu vede k jeho hromadění a nemožnosti jeho využití ve formě ATP. Pacient díky tomu trpí bolestí svalů a netoleruje svalovou námahu, po které dochází k rabdomyolýze s myoglobinurii, naopak, laktát se nezvyšuje (nedochází k anaerobní glykolýze, protože není substrát).
Typ II (Pompeho choroba) – viz kap. 432
3. Glykogenózy imitující hypertrofickou kardiomyopatii
Obě choroby se projevují hypertrofickou kardiomyopatií, oproti jiným typům mají výraznější elektrofyziologické abnormality (komorová preexcitace, poruchy vedení). Projevují se bolestmi na hrudníku, palpitacemi, synkopami a syndromem náhlé smrti. Léčbou volby u LAMP a nonkongenitální PRKAG 2 je transplantace srdce.
- Deficit LAMP2 (Danonova choroba, X dědičný deficit lysozomálního glykoproteinu LAMP2), k náhlé smrti dochází dříve (8 – 15 let), pacienti bývají často mentálně opoždění. Prognóza choroby je špatná.
- Deficit PRKAG2 (protein kinase AM-activated gamma 2, AD dědičná), k náhlé smrti dochází později (~ 33 let). Časté poruchy rytmu lze korigovat implantací pacemakeru. Prognóza choroby je lepší (mimo rychle progredující kongenitální formu, která je smrtící v časném dětství).
4. Vybrané poruchy metabolismu galaktózy
Metabolismus galaktózy – galaktóza je běžný sacharid potravy, který se do organismu dostává nejčastěji jako součást laktózy (glukóza a galaktóza vázaná 1,4 vazbou). Laktáza epitelu tenkého střeva uvolňuje z laktózy galaktózu, která se resorbuje a v játrech se rychle metabolizuje a proto se vůbec nedostává do moče. Metabolismus
- 1. Galaktóza se v játrech fosforyluje galaktokinázou na galaktóza-1-fosfát (Gal-1-P).
- 2. Na Gal-1-P se (stále v játrech) pomocí galaktóza-1-fosfát uridyltransferázy naváže uridylová skupina z UDP-glukózy za vzniku glukóza-1-fosfátu a UDP-galaktózy (UDP-Gal). Tato vazba má vysokou energii a vzniklá UDP-gal lze využít k syntéze glykolipidů, glykosaminoglykanů a glykoproteinů.
- 3. UDP-Gal se následně pomocí 4-epimerázy mění na glukózu.
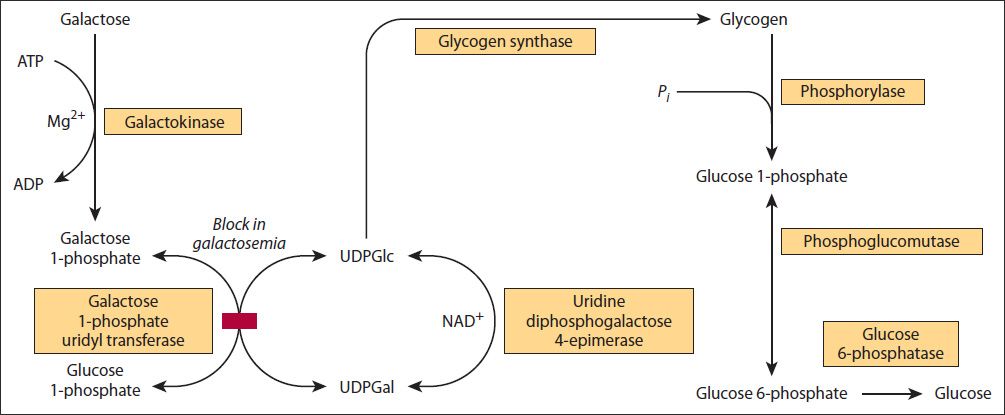
Galaktosémie
Etiologie – tato závažná choroba je způsobena deficitem galaktóza-1-fosfát uridyltransferázy (GALT) s incidencí 1 : 60 tisíc.
Patogeneze – zdravý novorozenec příjme > 40 % energie z laktózy, ze které vzniká galaktóza a glukóza. Při deficitu GALT se intermediální metabolit Gal-1-P hromadí a poškozuje ledviny, játra a mozek.
Klinický obraz
- po prvním nakojení novorozence může vzniknout zvracení, průjem a hepatomegalie s ikterem a selháním ledvin.
- poškození mozku se projeví častými poruchami řeči a někdy i poruchou rovnováhy a motorických funkcí.
- i přes dobrou léčbu je u 80 – 90 % postižených žen zjištěn hypergonadotropní hypogonadismus (ovariální selhání) s následnou neplodností.
CAVE Novorozenec s galaktosémií je ve zvýšeném riziku vzniku sepse z E. coli.
Diagnostika – zásadní je průkaz zvýšené koncentrace galaktikolu v moči a G1P v erytrocytech.
Terapie – základem je časná diagnóza (zaveden plošný screening ke zjištění její přítomnosti). Zásadním opatřením je eliminace galaktózy (a tedy i laktózy – mléka) z diety, což zvrátí jaterní a ledvinné selhání i růstový deficit, ale většinou již ne ovariální selhání.
Tělo není schopno metabolizovat laktózu a z ní vznikající galaktózu. Choroba se projeví 4. až 9. den po zahájení příjmu mléka. Hromadí se intermediární metabolit galaktóza-1-P a poškozuje mozek, játra (imituje akutní jaterní selhání), ledviny a ovaria u žen. Stav připomíná akutní sepsi s hepatorenálním selháním. Zásadou je doživotní vysazení mléka.
5. Vybrané poruchy metabolismu fruktózy
Metabolismus fruktózy – fruktóza je přijímána do organismu v medu a ovoci jako monosacharid, ale mnohem častěji jako disacharid sacharóza, která je ve střevě štěpena sacharázou na fruktózu a glukózu. Rychle se vstřebává střevem do portální krve a velmi rychle se vychytává v játrech. Všechna fruktóza (vstřebaná i endogenně produkovaná, např. jako meziprodukt glykolýzy) se zapojí do glykolýzy a to dvěma cestami o různé lokalizaci:
- 1. Ve svalu hexokináza fosforyluje fruktózu na fruktóza-1-fosfát (F-1-P), který již přímo vstupuje do glykolýzy. Takto se ovšem utilizuje jen malé množství fruktózy.
- 2. Daleko významnější je přeměna fruktózy v játrech:
- a. Fruktóza není substrátem pro jaterní hexokinázu, ale existuje zde specifický enzym fruktokináza, která dává vznik F-1-P.
- b. F-1-P je štěpen aldolázou B (jinou než v glykolýze) na:
- dihydroxyacetonfosfát, který vstupuje přímo do glykolýzy.
- glyceraldehyd, který je fosforylován specifickou kinázou na glyceraldehyd-3-fosfát, který opět vstupuje do glykolýzy.
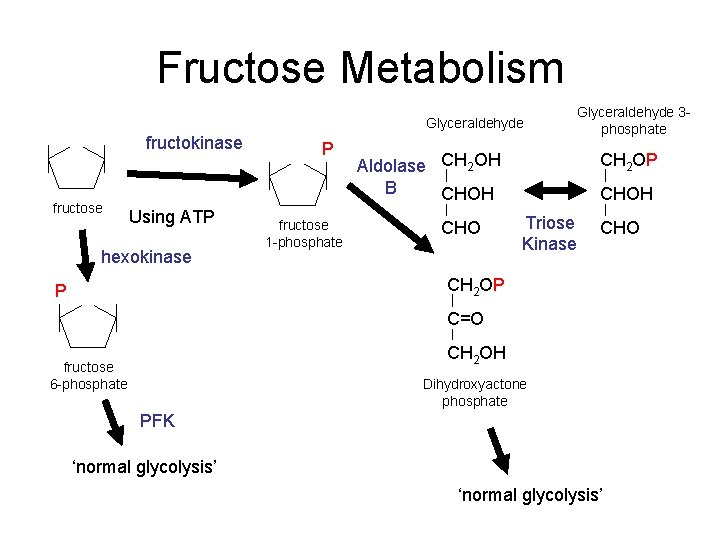
Deficit fruktokinázy (esenciální fruktózémie)
Benigní deficit fruktokinázy. Lze jej náhodně prokázat nálezem fruktózy v moči.
Deficit aldolázy B (vrozená intolerance fruktózy)
Při pozření fruktózy nebo sacharózy dochází ke zvracení s křečemi s příznaky selhání jater a ledvin. Zásadou je eliminovat fruktózu i sacharózu z potravy. Častá je současná celiakie.
Závažná choroba dětí, která je způsobena deficitem fruktóza 1,6-bisfosfát aldolázy. Zůstává asymptomatická, dokud pacient nepozře fruktózu nebo sacharózu (ovoce, sladké cereálie apod.). Pak vzniká zvracení s křečemi a letargií, hepatomegalie s ikterem. Laboratorně je elevace aminotransferáz i bilirubinu, hypoalbuminémie, prodloužení tvorby koagula (↑ clotting time) a porucha funkce proximálního tubulu. Pokud se nepřeruší přívod fruktózy, dochází ke vzniku hypoglykémií, selhání jater i ledvin a smrti. Podstatou léčby je eliminace všech zdrojů fruktózy, sacharózy a sorbitolu z potravy. Poté je prognóza choroby dobrá.
CAVE Často současná celiakie (> 10 %).
Deficit fruktóza-1,6-bisfosfatázy
Fruktóza-1,6-bisfosfatáza je zásadním regulačním enzymem glukoneogeneze. Při jeho dysfunkci se hromadí fruktóza-1,6-bisfosfát, který v nadbytečném množství vstupuje do glykolýzy. Při poklesu p.o. příjmu (horečka, gastroenteritida), kdy se za normálních okolností zvyšuje glukoneogeneze, se již od dětství projevuje hypoglykémií a laktátovou acidózou s hyperventilací a křečemi až kómatem. Na rozdíl od předchozího zůstává funkce ledvin i jater v normě. Při léčbě akutních epizod je nutná i.v. korekce hypoglykémie a acidózy. Jejich prevencí je vyvarování se lačnění a eliminace všech zdrojů fruktózy a sacharózy z potravy. K prevenci hypoglykémie lze užít např. kukuřičný škrob. Prognóza je dobrá a další vývoj v normě.