Úvod – lipoproteiny jsou komplexy lipidů a proteinů, které jsou zásadní pro transport cholesterolu (CH), triglyceridů (TG) a vitamínů rozpustných v tucích. Léčba dyslipidémií snižuje riziko aterosklerotických kardiovaskulárních chorob.
Klasifikace lipoproteinů – lipoproteiny jsou velké makromolekulární komplexy složené z lipidů a proteinů, které transportují špatně rozpustné lipidy tělními tekutinami (plazma, intersticiální tekutina, lymfa) mezi cílovými tkáněmi. Jsou tvořeny hydrofobním jádrem (TG, estery CH), obklopeným obalem z apolipoproteinů a hydrofilních lipidů (neesterifikovaný CH, fosfolipidy).

Plazmatické lipoproteiny jsou rozděleny na základě jejich hustoty do pěti hlavních tříd: chylomikrony, VLDL, IDL, LDL a HDL (very low, low, intermediate a high density lipoproteins). Lipidy mají nižší hustotu než voda, hustota a velikost lipoproteinové částice je proto dána obsahem lipidů v ní uloženého (na tuk nejbohatší chylomikrony jsou nejméně husté a největší, pro HDL platí opak).

Strukturální lipoproteinové proteiny (apolipoproteiny) jsou nutné pro správnou funkci i metabolismus lipoproteinové částice (aktivují enzymy, působí jako ligandy povrchových receptorů):
- ApoB jsou velké apolipoproteiny, které se dále dělí na apoB-48 (tvořené střevem, obsažené v chylomikronech) a apoB-100 (tvořené játry, obsažené ve VLDL, IDL, LDL).
- ApoA-I jsou obsažené ve všech HDL, tvořené střevy a játry.
- ApoA-II přítomny na cca 2/3 HDL částic.
- ApoC-I, C-II, C-III, E na povrchu lipoproteinů bohatých na TG.
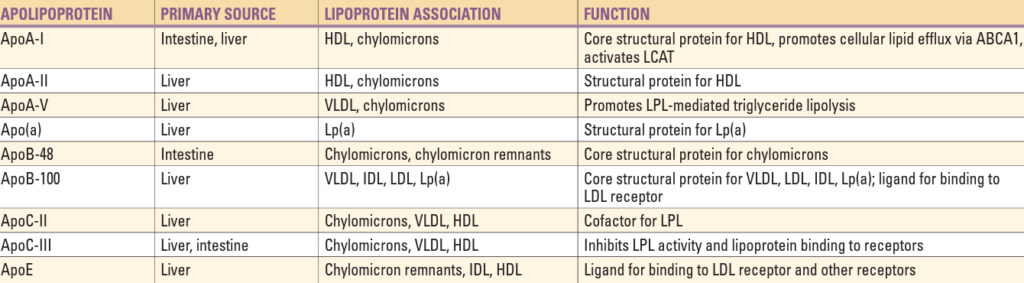
Transport lipidů lipoproteiny – lipidy jsou jednak transportovány ze střeva do periferních tkání a jater pomocí chylomikronů (exogenní metabolická cesta), jednak z jater do periferie (endogenní metabolická cesta). Naopak, CH z periferních tkání je do střeva a jater (pouze hepatocyty a enterocyty jej umí efektivně vylučovat z těla ven) transportován za pomocí HDL (reverzní transport CH).
a) Exogenní metabolická cesta (transport dietních lipidů ze střeva)
- 1. Hydrolýza TG z potravy lipázami uvnitř lumen střeva, následně absorpce CH, mastných kyselin (MK) a vitamínů rozpustných v tucích v proximální části tenkého střeva.
- 2. Esterifikace CH a retinolu na cholesteryl a retinyl estery. Mastné kyseliny s dlouhým řetězcem (LCFA, > 12 C) jsou inkorporovány do triglyceridů a spolu s apoB-48, CH, fosfolipidy a výše zmíněnými estery dávají vzniknout nascentním chylomikronům, které jsou secernovány do lymfy a cestou ductus thoracicus odchází do krve, kde intenzivně interagují s periferními tkáněmi předtím, než dosáhnou jater.
- 3a. Lipoproteinová lipáza (LPL) je enzym uchycený GPI (glykosylfosfatydilinositolovou) kotvou na proteinu GPIHBP1, nacházející se na povrchu endotelu tukové tkáně, kosterního svalstva a srdce. Jakmile se TG vázaný na chylomikron setká s LPL, dochází ke štěpení a uvolnění mastných kyselin (kofaktorem reakce s LPL je apoC-II), které jsou vychytávány adipocyty nebo myocyty a oxidovány za vzniku energie nebo reesterifikovány a uloženy ve formě TG.
- 3b. Některé uvolněné MK se vážou na albumin a jsou transportovány do ostatních tkání (zejména jater).
- 4. Po uvolnění MK dochází k výraznému zmenšení velikosti chylomikronů, hydrofobní jádro je hydrolyzováno (CH, fosfolipidy a apolipoproteiny jsou transportovány do HDL) se vznikem chylomikronových remnant.
- 5. Chylomikronové remnanty jsou rychle vychytávány z cirkulace játry procesem, jehož kofaktorem je apo-E.
- …TAG jsou inkorporovány do chylomikronů (apo B-48), přes lymfu jdou do krve a na periferii pomocí LPL (kofaktorem apoC-II) uvolňují MK poté jsou jako remnanty vychytávány játry (kofaktorem je apoE).
CAVE Celý proces je velmi rychlý, proto, pokud jsou chylomikrony nebo jejich remnanty přítomny v krvi i po 12 hodinovém lačnění je pravděpodobná porucha lipoproteinového metabolismu.
b) Endogenní metabolická cesta (transport lipidů z jater pomocí VLDL a LDL)
- 1. VLDL jsou podobné chylomikronům, ale místo apoB-48 obsahují apoB-100 a mají nižší poměr TG : CH. Jejich TG vznikají zejména esterifikací LCFA v játrech. TG z jater + CH estery + fosfolipidy + vitamin E + apoB-100 za účasti MTP (microsomal triglyceride transfer protein) dávají vznik nascentním VLDL.
- 2. Po uvolnění do plazmy VLDL přijímají více kopií apoE a apoC transferem z HDL.
- 3. TG z VLDL jsou hydrolyzovány LPL (podobně jako chylomikrony, zejména ve svalech, srdci a tukové tkáni) se vznikem VLDL remnant, které se po odpojení od LPL označují jako IDL (poměr TG a CH ≈ 1).
- 4a. V játrech dochází k vazbě LDL receptoru na apoE a endocytóze cca poloviny IDL.
- 4b. Zbytek IDL je modifikován (hydrolýza fosfolipidů i TG a odstranění všech apolipoproteinů mimo apoB-100) za pomocí jaterní lipázy (HL) za vzniku LDL. Z cirkulace je odstraněno cca 70 % LDL játry.
- 5. Lp(a) jsou lipoproteiny ve svém složení (proteiny a lipidy) podobné LDL, obsahují ale navíc apo(a), který je syntetizován i eliminován v játrech a na apo-B100 je vázán disulfidovou (-S-S-) vazbou.
…TG s LFCA, CH, apo-B100 za účasti MTP tvoří VLDL. Poté do plazmy a na periferii, kde předají pomocí LPL MK a stávají se IDL, které jdou do jater. 50 % internalizuje po vazbě na LDL receptor, 50 % je pomocí HL konvertováno na LDL, které předává CH periferním tkáním.

c) Metabolismus HDL a reverzní transport cholesterolu
- 1. Nascentní částice HDL jsou syntetizovány v játrech a střevu, kdy nově vytvořený apoA-I rychle shromažďuje fosfolipidy a neesterifikovaný CH z místa svého vzniku (játra, střevo) pomocí effluxu zajištěného membránovým proteinem ABCA1 (ATP-binding cassette protein A1).
- 2. Tímto dochází ke vzniku diskoidních HDL částic, které vážou další neesterifikovaný CH z buněk nebo ostatních lipoproteinů. Tento cholesterol je následně esterifikován pomocí plazmatického enzymu LCAT (lecitincholesterolacyltransferáza). CH estery jsou hydrofobní a pohybují se do jádra HDL částice, která se stává sférickou.
- 3. HDL-CH je poté transportován do jater:
- nepřímo pomocí CETP (cholesterylester transfer protein), který umožňuje směnu esterů CH za TG mezi HDL částicemi a lipoproteiny obsahujícími apo-B a zde odstraněn z cirkulace endocytózou pomocí LDL receptorů.
- přímo v játrech cestou povrchového receptoru hepatocytů SR-B1 (scavenger receptor class B1).
- 4. HDL částice poté v plazmě prodělávají rozsáhlou remodelaci spojenou se zvýšením obsahu fosfolipidů a TG. Ty se stávají substrátem pro jaterní i endoteliální lipázu, které poté fosfolipidy a TG hydrolyzují, což je spojeno s jejich zmenšením.
… v játrech a střevech se do HDL se pomocí ABCA1 pumpuje cholesterol se vznikem diskoidní formy HDL, která váže další volný cholesterol, který se uvnitř pomocí LCAT esterifikuje za vzniku sférické formy HDL, která jde následně do jater.
I. Choroby spojené se zvýšením cholesterolu a triglyceridů
Poruchy lipoproteinového metabolismu se nazývají dyslipidémie. Na jejich vzniku se většinou podílí genetické predispozice (často polygenní) spolu s faktory prostředí (životní styl, léky, zdravotní stav). Patofyziologicky lze dyslipidémie rozdělit do pěti skupin na dyslipidémie způsobené…
I.1. …nadměrnou produkcí VLDL
Při zvýšení hladiny VLDL bohaté na triglyceridy) je obvykle ↑ TG, ↓ HDL-CH, různá hladina LDL-CH, ale ↑ apo B a jsou obvykle přítomny i ostatní znaky metabolického syndromu, které mohou s ostatními faktory ovlivňovat vlastní syntézu VLDL (obezita, inzulinová rezistence, vysokosacharidová dieta, alkoholismus, exogenní estrogeny i genetické predispozice).
I.1a Sekundární příčiny
Vysokosacharidová dieta – zvýšené množství sacharidů v potravě („západoevropská strava“) → v játrech konverze na volné mastné kyseliny (FFA) → esterifikace MK se vznikem triglyceridů → VLDL.
Alkohol – pravidelná konzumace alkoholu → inhibice jaterní oxidace FFA → stimulace jaterní syntézy TG → VLDL. Dalším projevem je elevace HDL-CH. Proto ↑ triglyceridů i ↑ HDL-CH = podezření na alkoholismus.
Obezita a inzulínová rezistence – zvýšené množství tukové hmoty vede ke zvýšení FFA transportovaných do jater, dále inzulinová rezistence s hyperinzulinémií zvyšují jejich jaterní syntézu s reesterifikací na TGa zvýšením produkce VLDL v játrech. Hyperinzulinémie dále snižuje aktivitu LPL se snížením katabolismus VLDL a chylomikronů.
Nefrotický syndrom – příčina zvýšení VLDL není plně objasněna, souvisí pravděpodobně s hypoalbuminémií a následnou proteosyntézou. Efektivní léčba nefropatie vede obvykle k normalizaci lipidogramu.
Cushingův syndrom – nadbytek glukokortikoidů vede ke zvýšení hladiny VLDL a TG a snížení hladiny HDL-CH.
I.1b Primární příčiny
Familiární kombinovaná hyperlipidémie (FCHL) – incidence choroby je 1: 100 – 200 (cca 20 % pacientů s manifestací ICHS před 60. rokem života trpí FCHL). Může se manifestovat i v dětství, plný projev choroby je obvykle až v dospělosti. Společným znakem je nadprodukce VLDL játry, konkrétní metabolický defekt není ale zcela objasněn (dědičnost je typicky polygenní). Typickým nálezem je zvýšení LDL nebo VLDL (tedy i TG) nebo obojí (všechny tři fenotypy se mohou u jednoho pacienta střídat v čase s ohledem na aktuální dietu, tělesnou hmotnost, fyzickou aktivitu a inzulínovou rezistenci), což lze prokázat i v úzkém příbuzenstvu pacienta (spolu s projevy předčasné aterosklerózy v rodinné anamnéze), dále zvýšení apoB, které nekoreluje s hladinou LDL cholesterolu, což odpovídá přítomnosti malých, denzních LDL částic. Hladina TG je 2 – 6,5 mmol/l, celkového CH 5 – 10 mmol/l a HDL-CH ˂ 1,0 mmol/l u mužů a ˂ 1,3 mmol/l u žen. Pacienti by měli být intenzivně léčeni, základem jsou režimová opatření (snížení příjmu jednoduchých cukrů, aerobní cvičení, redukce tělesné hmotnosti) spolu s farmakoterapií (na základě dominantního fenotypu statiny nebo fibráty nebo oboje).
Lipodystrofie – lokálně nebo i celkově je porušena schopnost tukové tkáně hromadit tuk, který je tak vychytáván jinými tkáněmi, zejména játry. Často je spojena s inzulínovou rezistencí a ↑VLDL a chylomikronů, díky zvýšení syntézy MK a snížení clearance lipoproteinových částic bohatých na TG. Pacienti s globální lipodystrofií jsou extrémně vzácní, chybí jim podkožní tuková tkáň a trpí výraznou inzulínovou rezistencí, deficiencí leptinu a výrazně akumulují TG v četných tkáních včetně jater. U některých pacientů bylo terapeuticky účinné podávání leptinu. Parciálnílipodystrofie je mírně častější a může být způsobena mutací několika genů, nejčastěji laminu A. Může se projevovat akumulací podkožního tuku v oblasti trupu v kontrastu s jeho chyběním na končetinách a hýždích. Dalším projevem je inzulínová rezistence, diabetes mellitus 2. typu, steatóza jater a dyslipidémie (zvýšení TG a CH, které je obtížně léčebně zvládnutelné). U pacientů je vzhledem k vysokému kardiovaskulárnímu riziku nutná intenzivní hypolipidemická léčba

I.2. …porušenou lipolýzou chylomikronů a VLDL
LPL je klíčovým enzymem, který hydrolyzuje TG v chylomikronech a VLDL. Do extracelulárního prostoru je produkován adipocyty, myocyty, kardiomyocyty a makrofágy a poté je transportován do subendotelu cév pomocí GPIHPB1. Jedinci s porušenou funkcí LPL mají ↑ TG, ↓ HDL-CH a normální hladinu LDL-CH i apo-B, dále obvykle zvýšenou hladinu VLDL a inzulinovou rezistenci.
I.2a Sekundární příčiny
Obezita a inzulínová rezistence – mimo zvýšenou produkci VLDL (viz výše) dochází i ke snížení aktivity LPL, pravděpodobně díky redukci transkripce LPL ve tkáních a zvýšení produkce LPL inhibitoru apoC-III v játrech s následným vznikem dyslipidémie.
I.2b Primární příčiny
Familiární chylomikronémie – podstatou choroby je snížení aktivity:
- LPL – AR dědičnost, prevalence homozygotů 1 : 1 miliónu, heterozygoti trpí mírnou až střední hypertriglyceridémií.
- apoC-II – kofaktor LPL, také AR dědičnost, prevalence homozygotů ještě nižší než u deficitu LPL, heterozygoti hypertriglyceridémií netrpí.
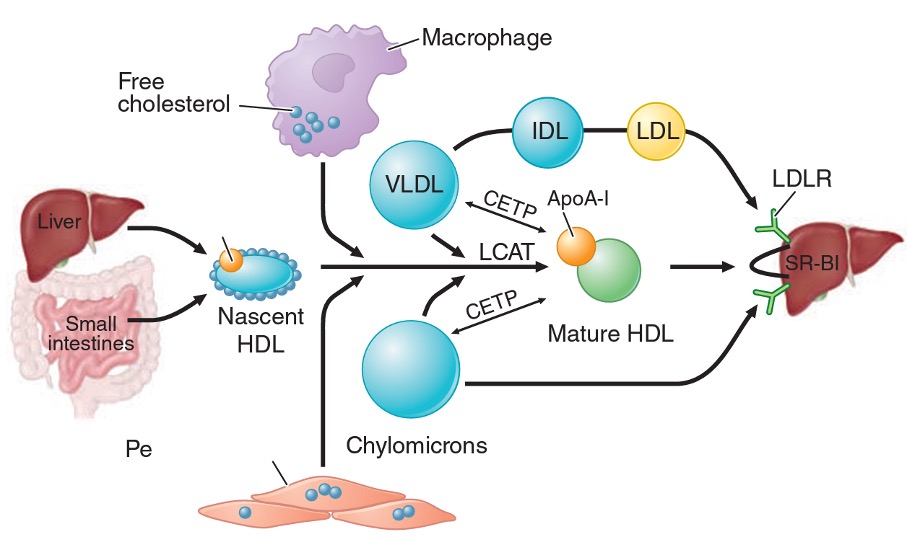
Nedostatečná aktivita LPL nebo kofaktoru apoC-II vede k hromadění VLDL a chylomikronů. Choroba se projevuje již v dětství opakovanými záchvaty akutní pankreatitidy, při vyšetření očního pozadí lze prokázat opalescentní krevní cévy (lipemia retinae), běžné jsou eruptivní xantomy (malé, žlutobílé papuly na zádech, hýždích a extenzorových plochách končetin), které nebývají bolestivé, ale mohou svědit. Ve fyzikálním nálezu bývá hepatosplenomegalie (zvýšený příjem chylomikronů RE systémem). Choroba obvykle nebývá spojena s předčasnou manifestací ICHS. Situace je ještě obtížnější v těhotenství, kdy ke zvýšené tvorbě VLDL dochází fyziologicky.
CAVE U některých pacientů mohou tyto nálezy chybět.
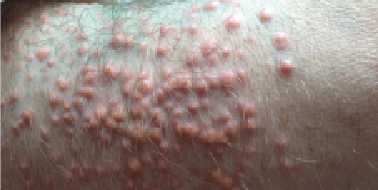
V laboratorním nálezu dominuje zvýšení plazmatické hladiny chylomikronů (VLDL i LDL bývá vyšší, ale méně výrazně), což vede k vytvoření krémovitého supernatantu na povrchu plazmy ponechané několik hodin stát při teplotě 4°C. Laboratorně je přítomna těžká hypertriglyceridémie (často ˃ 11 mmol/l), hladina CH je také vyšší, ale ne až tak markantně. Diagnóza je dána enzymatickým vyšetřením ve specializované laboratoři. Aktivita LPL bývá u obou defektů snížená, u defektu apoC-II se lipolytická aktivita upraví přidáním „zdravé“ plazmy (zdroj apoC-II). Ke konfirmaci diagnózy lze využít genetické vyšetření.
Při léčbě je nutné dodržovat režimová opatření (příjem tuku v potravě < 15 g/den, suplementace vitamínů rozpustných v tucích, v některých případech podávání MK se středním řetězcem, které jsou absorbovány přímo do portálního oběhu), to vše za dohledu dietního specialisty. Pokud tato opatření nejsou účinná, lze zkusit rybí olej. U pacientů s deficitem apoC-II je v akutním případě indikováno podání plazmy. K použití je schválena genová terapie pomocí preparátu alipogen tiparvovec, který spočívá v mnohočetném intramuskulárním podání LPL virovým vektorem, což vede ke zvýšené expresi LPL myocyty kosterního svalstva.
ApoA-V deficience – apoA-V usnadňuje interakci chylomikronů a VLDL s LPL a jejich hydrolýzu. Při hypofunkční mutaci APOA5 alel dochází k hyperchylomikronémii, heterozygoti se pravděpodobně podílí na polygenní variantě hypertriglyceridémie.
Deficit GPIHBP1 – homozygoti trpí těžkou hypertriglyceridémií, protože vázne transport LPL na povrch cévního endotelu. Frekvence choroby nebyla stanovena, ale předpokládá, se že je velmi raritní.
Familiární hypertriglyceridémie (FHTG) – ↑ TG bez jasné sekundární příčiny (tuto je vždy nutné vyloučit), hladiny LDL i apoB bývají normální nebo snížené (zhoršení konverze lipoproteinů bohatých na TG na LDL), proto tato choroba nebývá spojena se zvýšeným KV rizikem. Hlavním rizikem FHTG je vznik akutní pankreatitidy. V léčbě je vždy nutné začít s dietou s přísným omezením tuku, pokud je hladina TG > 5 mmol/l je vhodné zahájení léčby fibráty nebo rybím olejem.
I.3 …porušeným jaterním příjmem lipoproteinů obsahujících apo-B
Dochází k zhoršení příjmu zejména LDL lipoproteinů. Asi nejzásadnější režimová chyba vedoucí ke snížení aktivity LDL receptoru je zvýšený příjem trans-mastných kyselin.
I.3a Sekundární příčiny
Hypotyreoidismus – vede k porušení funkce LDL receptorů (hormony štítné žlázy zvyšují jejich aktivitu).
Chronická renální insuficience – pacienti s chronickým postižením ledvin mají lehké ↑ TG i LDL cholesterolu díky prodloužení clearance VLDL.
Jaterní choroby – hepatopatie (hepatitidy, léky, alkohol) vedou ke zvýšení hladiny VLDL a triglyceridů. Naopak, těžké jaterní poruchy mají za následek pokles produkce lipoproteinů a tedy i snížení plazmatické hladiny CH i TG. Většina CH je vylučována do žluči, proto je cholesteáza spojena s výraznou hypercholesterolémií se vznikem eruptivních i planárních xantomů.
Estrogeny – jejich podání vede ke zvýšení VLDL i HDL se vznikem hyperTG i hyperHDL.
I.3b Primární příčiny
Familiární hypercholesterolémie
Epidemiologie – četnost heterozygotů je 1: 250 až 500, u homozygotů 1: 500 tisíc až 1 milión.
Etiologie – dědičnost choroby je AD, nejčastější příčinou je:
- mutace genu pro LDL receptor (LDLR)
- méně často pro APOB – apoB100 je kritický pro vazbu LDL částice na LDL receptor nebo
- PCSK9 – proprotein convertase subtilisin/kexin typ 9 (PCSK9) je protein, který se váže na LDL receptor v játrech a indukuje jeho internalizaci a disociaci (normálně se vrací zpět na buněčný povrch a čeká na další LDL lipoprotein). PCSK9 tak snižuje clearance LDL lipoproteinů.
Klinický obraz
- heterozygoti – hladina LDL cholesterolu je 5 – 10 mmol/l, je časnější nástup arcus cornealis a/nebo šlachových xantomů (zejména dorzum rukou a Achillovy šlachy). Vždy by měly být vyloučeny sekundární příčiny dyslipidémie, zejména hypotyreoidismus, nefrotický syndrom a cholestáza. K diagnóze mohou pomoci Dutch kritéria (https://www.mdcalc.com/dutch-criteria-familial-hypercholesterolemia-fh). Základem léčby jsou statiny, při nedosažení cílových hodnot je indikován ezetimib, popřípadě PCSK9 inhibitory.
- homozygoti – přítomna mutace obou alel LDL receptoru. LDL cholesterol dosahuje hodnot 10 až > 25 mmol/l, zatímco TG bývají normální. Již od dětství jsou přítomny xantomy rukou, kloubů, loktů, kolen a hýždí. Zásadním a devastujícím důsledkem je extrémní akcelerace aterosklerotického cévního postižení. Homozygoti s absencí LDL receptorů nemají léčebnou odpověď na statiny, PCSK9 inhibitory nebo inclisiran (malé interferující RNA, které inhibují PCSK9), u pacientů s jejich částečnou expresí je alespoň částečnou odpověď na terapii. Alternativní léky, které redukují jaterní produkci VLDL a tak i LDL cholesterolu jsou:
- inhibitor microsomal TG transfer protein (MTP) lomitapid
- inhibitor apoB mipomersen
Sitosterolémie – vzácné AR dědičná mutace ATP-binding casette proteinu ABCG5 a ABCG8, který funguje jako transportér sterolů (rostlinný sitosterol a campesterol a živočišný cholesterol) přes biliární membránu hepatocytů do žluče a přes membránu enterocytů do střeva. U zdravých jedinců je v tenkém střevě resorbováno < 5 % rostlinných sterolů z potravy a malé množství, které vnikne do cirkulace je následně vyloučeno do žluči a následně střeva – koncentrace ve tkáni je tedy velmi nízká. U sitosterolémie je zvýšena resorpce sterolů střevem a snížena jejich sekrece do žluče s následným zvýšením koncentrace rostlinných sterolů a cholesterolu v plazmě i tkáních. Pacienti mají mimo typické příznaky hypercholesterolémie (xantomy, akcelerace aterosklerózy) i nález anisocytózy, poikilocytózy erytrocytů a megatrombocytů se vznikem hemolýzy a splenomegalie. Podezření na sitosterolémii by mělo vzniknout u pacientů s těžkou hypercholesterolémií bez pozitivní rodinné anamnézy a se selháním statinové terapie. K potvrzení diagnózy slouží genetické vyšetření. Základem terapie je dieta, cholestyramin a ezetimib. U heterozygotů je nález pouze střední hypercholesterolémie.
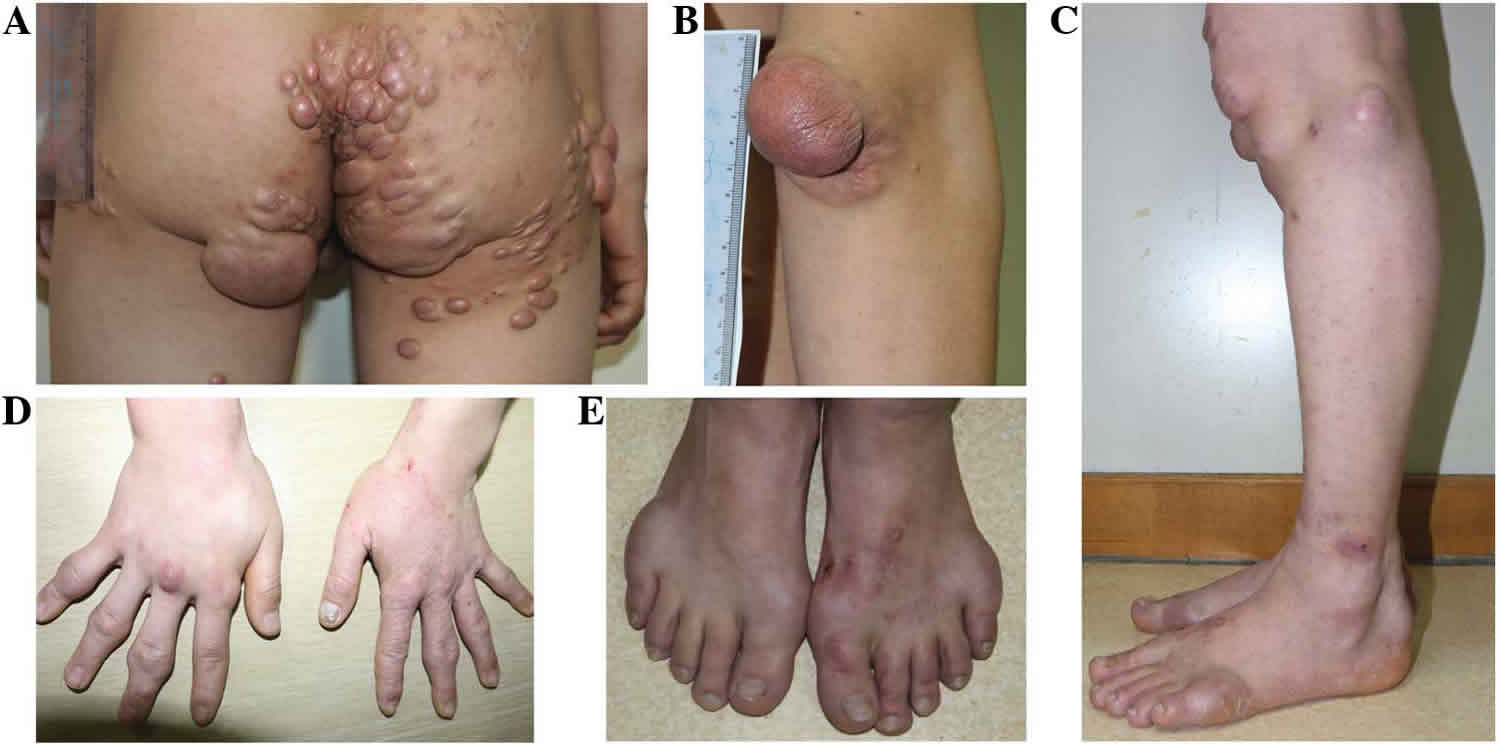
Familiární dysbetalipoproteinémie (FDBL) – většinou AR dědičná porucha vazby apoE (nejčastěji apoE2) na své receptory. Dochází k akumulaci chylomikronových remnant, VLDL remnant i IDL v plazmě a díky tomu i smíšené dyslipidémie (hyperCH i hyperTG). Gen apoE má možnost polymorfismů:
- apoE3 – nejčastější
- apoE4 – heterozygoti i homozygoti mají zvýšené riziko vzniku Alzheimerovy demence.
- apoE2 – mají nižší afinitu k LDL receptoru, proto u hetero- i homozygotů je nižší clearance lipoproteinů obsahujících apoE2 (chylomikronové remnanty, IDL) s jejich následným hromaděním. Důsledkem je smíšená dyslipidémie.
CAVE Homozygotů apoE2 je 0,5 % celkové populace, ale jen menší část z nich má typickou smíšenou dyslipidémii. Je proto pravděpodobně potřeba ještě další faktor, který spouští manifestaci FDBL (např. dieta s vysokým obsahem tuků, diabetes mellitus, obezita, hypotyreoidismus, postižení ledvin, deficit estrogenů, alkoholismus, deficit estrogenů, některé léky apod.).
FDBL se při své manifestaci projevuje vznikem tuberoeruptivních xantomů (shluky malých xantomů na loktech, kolenech a hýždích) a palmárních xantomů (žlutooranžové zbarvení palmárních a zápěstních rýh).
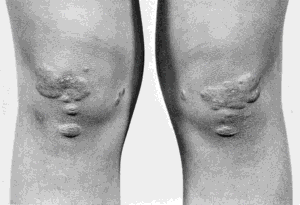

Definitivní diagnóza je dána nálezem vysokých hladin remnantních chylomikronů a IDL a/nebo genetickým průkazem genotypu apoE2/apoE2.
Ke stanovení LDL cholesterolu nelze použít Friedwaldovu formuli, protože v částicích VLDL u pacientů s FDBL je nedostatek TG a naopak zvýšené množství cholesterolu.
Hlavním rizikem FDBL je zvýšení rizika kardiovaskulárních chorob, proto je ji třeba agresivně léčit. Nutná je redukce hmotnosti, dieta s omezením tuků i cholesterolu, vyvarování se alkoholu a statiny, při nedostatečném účinku ezetimib, fibráty a PCSK9 inhibitory.
I.4. …nízkou hladinou lipoproteinů obsahujících apo-B
Abetalipoproteinémie – vzácná AR dědičná hypofunkční mutace genu kódujícího microsomal TG transfer protein (MTP), který zajišťuje transport lipidů do nascentních chylomikronů ve střevě a VLDL v játrech, proto je plazmatická hladina CH i TG velice nízká a VLDL, LDL a apo-B nelze detekovat. Hlavní příčinou klinické manifestace choroby je malabsorpce vitamínů rozpustných v tucích (zejména vitamínu E, méně A a K – vitamín E je normálně transportován ze střeva v chylomikronech a z jater ve VLDL). Choroba se projevuje od časného dětství:
- opoždění růstu, steatorea díky malabsorpci tuků.
- neurologicky – zpočátku ztráta hlubokých šlachových reflexů, poté propriocepce, dysmetrie, ataxie až spastická chůze ve věku cca 30 roků.
- vizuální – pigmentová neuropatie následovaná šeroslepostí až téměř slepotou.
Základem léčby je nízkotučná a vysokokalorická dieta a bohatá suplementace vitamíny rozpustných v tucích (zejména vitamínu E).
Familiární hypobetalipoproteinémie – situace nízkého celkového i LDL cholesterolu (včetně apo-B) s postižením tvorby chylomikronů ve střevě a VLDL v játrech. Dochází ke zvýšenému clearance VLDL a lipoproteinů, které z nich pochází, s následnou retencí tuků v játrech se vznikem NAFLD. Extrémně vzácní homozygoti vedou k obrazu podobnému abetalipoproteinémii.
Familiární kombinovaná hypolipidémie – gen pro angiopoietin-like 3 (ANGPTL3) kóduje stejnojmenný protein produkovaný játry, který inhibuje LPL a tak prodlužuje clearance lipoproteinů bohatých na TG. Hypofunkční mutace tak zvyšuje aktivitu LPL a snižuje plazmatickou koncentraci TG a zároveň i snižuje riziko ischemické choroby srdeční.
PCSK9 deficience – hypofunkční mutace pro PCSK9 výrazně snižuje hladinu LDL cholesterolu a tak i KV riziko (u heterozygotů je LDL o cca 30 – 40 % nižší oproti osobám bez mutace, u homozygotů je LDL většinou < 0,5 mmol/l).
II. Choroby spojené s nízkou hladinou HDL
Nízká hladina HDL CH je nezávislým prediktivním faktorem zvýšeného KV rizika. Metabolismus HDL je silně ovlivněn metabolismem TG a zejména obezitou a inzulínovou rezistencí, naopak k jeho zvýšení pomáhá dostatečná fyzická aktivita.
II.1 Vrozené příčiny nízké koncentrace HDL
Mutace genu pro apo-AI – delece tohoto genu se projeví absencí HDL lipoproteinů v plazmě. Volný cholesterol působí opacity v čočce a plantární xantomy.
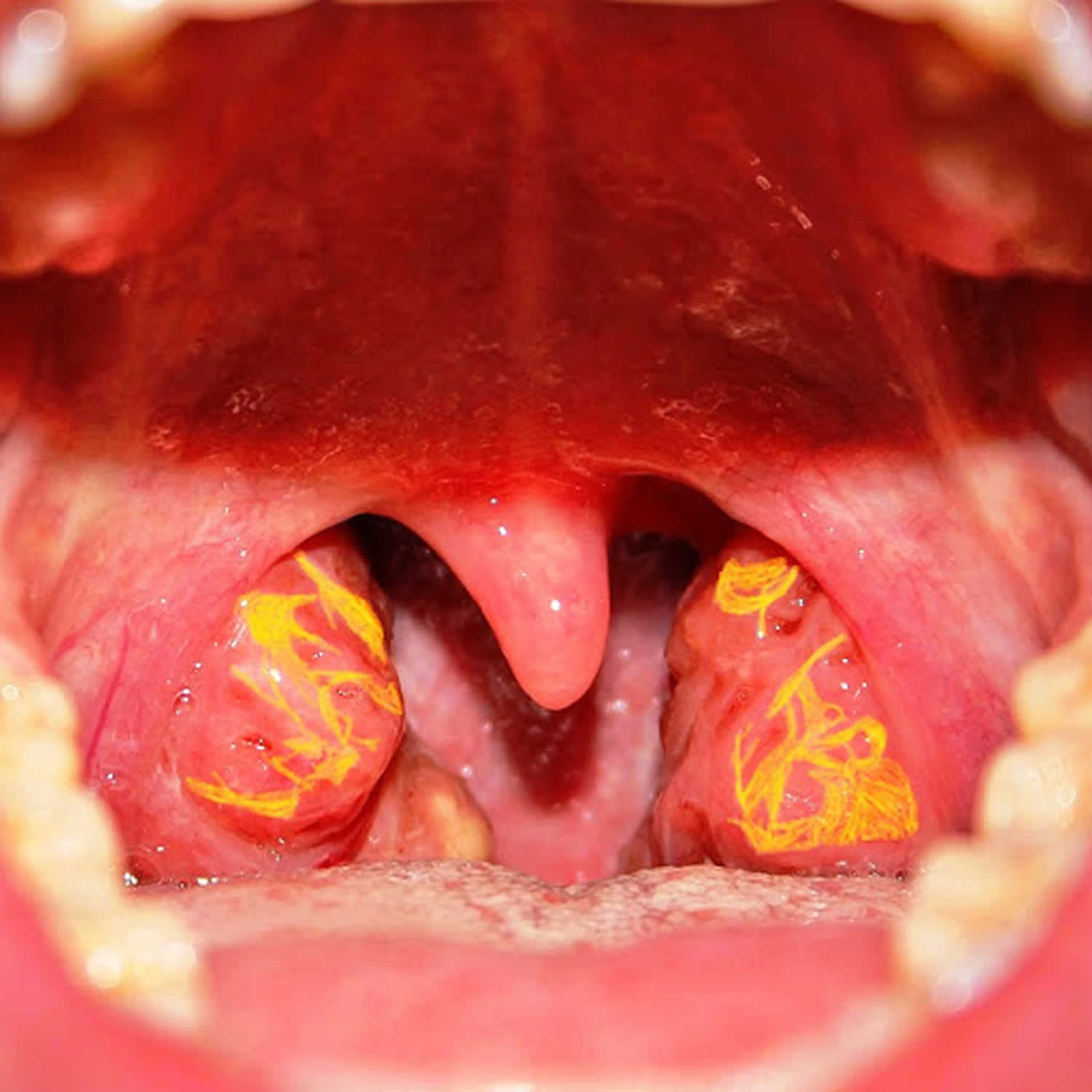
Tangierova choroba – vzácná AR dědičná choroba způsobená mutací ABCA1, buněčného transportéru, který usnadňuje přesun CH a fosfolipidů z buněk na apo-A1. Při jeho nepřítomnosti jsou špatně lipidované apo-AI ihned odstraňovány z cirkulace, důsledkem je nízká plazmatická koncentrace apo-AI i HDL CH (< 0,15 mmol/l). Nadbytečný cholesterol se akumuluje ve tkáních RES, zejména v játrech (hepatosplenomegalie) a patrových tonzilách (zvětšené šedožluté až oranžové tonzily – PATOGNOMONICKÉ).
Je mírné zvýšení KV rizika, ale ne tak výrazně jak bychom očekávali při tak nízkých hladinách HDL, pravděpodobně díky i současnému poklesu LDL cholesterolu.
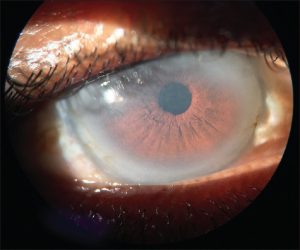
Familiární deficit LCAT – vzácná AR dědičná ztrátová mutace LCAT. Nedochází k esterifikaci CH za vzniku CH esterů, dochází tak k porušené maturaci HDL částic (z diskoidních nevznikají sférické) a rychlému clearance apo-AI se vzestupem frakce volného cholesterolu z celkového na cca 70 % (norma je cca 25 %). Choroba není spojena s vyloženě předčasnou manifestací aterosklerózy. Existují dvě formy:
- fish-eye disease – částečný deficit LCAT. Projevuje se progresivními opacitami v rohovce a velmi nízkou koncentrací HDL cholesterolu.
- klasický deficit LCAT – kompletní chybění LCAT. Předchozí + hemolytická anémie a renální insuficience.
Primární hypoalfalipoproteinémie – výrazný deficit HDL při vyloučení deficitu LCAT i Tangierovy choroby. Příčinou je pravděpodobně urychlený katabolismus HDL.
Diagnostika dyslipidémií
Odběr lipidogramu by měl být proveden po nejméně 12 hodinovém nočním lačnění. Cholesterol a triglyceridy jsou měřeny přímo, LDL cholesterol je dopočítáván Friedwaldovou formulí:
LDL = celkový cholesterol – (TG/5) – HDL
CAVE Nepřesné při TG > 4,5 mmol/l!
Terapie dyslipidémií
Léčba těžké hyperTG k prevenci vzniku akutní pankreatitidy
Obecně, intenzivní léčba by měla být zahájena při TG > 5,5 mmol/l:
- 1. Změna životního stylu – základem je redukce příjmu alkoholu a jednoduchých cukrů, redukce hmotnosti (za pomoci dietního specialisty v těžkých případech i bariatrické chirurgie).
- 2. Farmakoterapie – pokud i přes úpravy životního stylu zůstává hladina TG > 5,5 mmol/l, je vhodné zahájení terapie:
- fibráty – agonisté PPARα (nukleární receptory, které stimulují příjem TG játry, zvyšují jejich β-oxidaci i syntézu apo-CIII, která inhibuje LPL). Zejména v kombinaci se statiny mohou způsobovat myopatii.
- ω3 mastné kyseliny (n-3 PUFA) – nejčastěji eikosapentaneová a dokosahexaenová kyselina jsou obsaženy v rybím oleji a snižují hladinu TG.
Léčba těžké hyperCH k prevenci kardiovaskulárních chorob
Cílové hladiny LDL cholesterolu závisí dle guidelines ESC na celkovém KV riziku:
Kategorie nízkého rizika – cílové LDL < 3 mmol/l – pouze zdánlivě zdraví lidé, kteří dosahují hodnoty SCORE2: věk < 50 let – pod 2,5 %, věk 50 – 70 let – pod 5 %, věk ≥ 70 let – pod 7,5 %.
Kategorie středního rizika – cílové LDL < 2,6 mmol/l (nebo redukce o > 50 % oproti výchozí hodnotě) – zdánlivě zdraví lidé, kteří dosahují hodnoty SCORE2 odpovídajícím nízkému riziku nebo
- diabetes mellitus II. typu – pacienti s dobře kontrolovaným diabetem < 10 let, bez ostatních rizikových faktorů KVR a bez orgánových manifestací diabetu.
Kategorie vysokého rizika – cílové LDL < 1,8 mmol/l (nebo redukce o > 50 % oproti výchozí hodnotě) – zdánlivě zdraví lidé, kteří dosahují hodnoty SCORE2: věk < 50 let 2,5 – 7,5 %, věk 50 – 70 let 5 – 10 %, věk ≥ 70 let 7,5 – 15 % nebo
- diabetes mellitus II. typu – pacienti bez ostatních rizikových faktorů KVR a bez orgánových manifestací diabetu, které nesplňují kritéria středního, ale ani velmi vysokého rizika (např. dobře kontrolovaní, jinak zdraví diabetici > 10 let trvání).
- chronická renální insuficience – CKD IIIbA1 nebo IIIaA2 nebo IIA3.
- výrazná dyslipidémie.
Kategorie velmi vysokého rizika – cílové LDL < 1,4 mmol/l (nebo redukce o > 50 % oproti výchozí hodnotě) – zdánlivě zdraví lidé, kteří dosahují hodnoty SCORE2: věk < 50 let ≥ 7,5 %, věk 50 – 70 let ≥ 10 %, věk ≥ 70 let ≥ 15 , nebo
- diabetes mellitus II. typu
- + prokázané mikrovaskulárním onemocněním nejméně ve 3 různých lokacích (např. retinopatie, neuropatie, nefropatie).
- + CKD IIIb nebo IIIaA2 nebo jakékoliv A3.
- chronická renální insuficience – CKD IIIbA2+3, IV a V.
- anamnéza kardiovaskulárního onemocnění.
Formy terapie:
- 1. Změna životního stylu – základem je redukce tělesné hmotnosti, omezení cholesterolu a nasycených mastných kyselin (zejména v trans konfiguraci) v dietě, pravidelná fyzická aktivita k zásadnímu poklesu LDL cholesterolu nevede (snižuje HDL a celkové KV riziko).
- 2. Farmakoterapie
- statiny – inhibují HMG-CoA reduktázu (klíčový enzym biosyntézy CH). Míra redukce plazmatické koncentrace LDL je interindividuálně velice rozdílná, nicméně následující zdvojnásobení dávky statinu vede k dalšímu poklesu LDL o cca 6 %. Statiny v závislosti na dávce částečně snižují i TG a vedou k vzestupu HDL o cca 5 – 10 %. V prevenci a léčbě kardiovaskulárních chorob je preferován atorvastatin a rosuvastatin. Mohou mít některé nežádoucí účinky:
- myopatie až rhabdomyolýza – poměrně vzácná komplikace, riziko se zvyšuje při vyšším věku, fragilitě, onemocnění ledvin a při požití léků zvyšujících jejich účinek formou interakcí. K posouzení závažnosti je vhodné odebrat CK.
- zvýšení jaterních testů (ALT, AST).
- zvýšení rizika vzniku diabetes mellitus II. typu o cca 10 % (toto riziko je bohatě vyváženo poklesem KV rizika).
- ezetimib – CH je v lumen tenkého střeva (1/3 pochází z potravy a 2/3 ze žluče) absorbován pomocí transportního proteinu NPC1L1. Tento transportér je inhibován pomocí ezetimibu a absorpce CH tak klesá o 60 %, což vede až k 18 % redukci plazmatické hladiny LDL cholesterolu a tento efekt se dále zvyšuje v kombinaci se statinem.
- vazače žlučových kyselin – vážou žlučové kyseliny a brání jejich reabsorpci v ileu. K udržení jejich poolu je nutná v játrech jejich zvýšená syntéza. Primárně snižují LDL cholesterol, ale mohou vést i ke zvýšení TG (jsou tedy nevhodné u pacientů s výraznější hyperTG před zahájením léčby). Hlavními nežádoucími účinky jsou nadýmání a zácpa. Mezi preparáty patří cholestyramin, colestipol a colesevelam. Jsou vhodné do kombinace se statiny a ezetimibem
- PCSK9 inhibitory – protilátky, které se vážou na cirkulující PCSK9 a brání jeho interakci s LDL receptorem a následně i jeho internalizaci a degradaci. Jejich podání vede ke snížení jak hladiny LDL o 50 – 60 %, tak KV rizika. Jsou vhodné jako léčba druhé linie při intoleranci statinu a/nebo ezetimibu za podmínek nedosažení cílové hladiny LDL. Zatím jsou dostupné alirocumab a evolocumab.
- specializovaná léčba u homozygotní FH – léky které redukují jaterní produkci VLDL a tak i LDL cholesterolu. Jsou vhodné při nedostatečném efektu PCSK9 inhibitorů:
- inhibitor microsomal TG transfer protein (MTP) lomitapid
- inhibitor apoB mipomersen
- inclisiran – lék schválen 12/2020 EMA k terapii FH. Jde o malou interferující RNA, která brání translaci a syntéze PCSK9.
- statiny – inhibují HMG-CoA reduktázu (klíčový enzym biosyntézy CH). Míra redukce plazmatické koncentrace LDL je interindividuálně velice rozdílná, nicméně následující zdvojnásobení dávky statinu vede k dalšímu poklesu LDL o cca 6 %. Statiny v závislosti na dávce částečně snižují i TG a vedou k vzestupu HDL o cca 5 – 10 %. V prevenci a léčbě kardiovaskulárních chorob je preferován atorvastatin a rosuvastatin. Mohou mít některé nežádoucí účinky:
- 3. LDL aferéza – indikována při neúspěchu redukce LDL cholesterolu na požadovanou úroveň pomocí farmakoterapie. Pacientova plazma prochází kolonou, kde jsou z ní selektivně odstraněny LDL částice. Měla by být zvážena u pacientů s ICHS, kteří mají LDL cholesterol nad cca 4 – 5 mmol/l.
2019 ESC Guidelines k léčbě dyslipidémií: https://academic.oup.com/eurheartj/article/41/1/111/5556353