Definice – většinou vrozená porucha absorpce železa ze střeva, která vede k jeho excesivnímu ukládání do různých orgánů s jejich následnou dysfunkcí. Skladovací pigment se nazývá hemosiderin a přítomnost barvitelného železa v tkáních hemosideróza.
Epidemiologie – u vrozené hemochromatózy je prevalence regionálně závislá (nejčastěji v Severní Evropě s prevalencí homozygotů 1: 200 – 300 a frekvencí mutace 1:10). 70 % pacientů má první symptomy ve věku 40 – 60 let (první manifestace před 20. rokem života je raritní). Ostatní formy jsou vzácné, postihují i mladé lidi a všechny rasy.
Etiologie a patogeneze – celkový obsah železa v organismu je normálně 3 – 4 gramy (udržováno vyrovnanou střevní resorpcí a sekrecí, cca 1 mg/den u mužů, 1,5 mg/den u žen). Při hemochromatóze je celkový obsah železa 20 gramů a více, se střevní absorpcí 4 mg denně a více. Při akumulaci železa dochází ke zvyšování saturace transferinu a zvýšení zásobního železa (feritinu). Železo se hromadí zejména v játrech a pankreatu (až 100x vyšší než norma), srdci (až 25x vyšší než norma) a hypofýze (často příčina hypofyzárního hypogonadismu).
- 1. Hereditární hemochromatóza – AR dědičná mutace HFE genu (chromozóm 6p, identifikován v roce 1996). Nejčastěji:
- C282Y – záměna G → A vede k záměně cystein → tyrosin na pozici 282 (u 85 – 90 % pacientů s hereditární hemochromatózou, ale ve Středozemí, např. jižní Itálii pouze u 60 % případů).
- H63D – záměna histidin → aspartát na pozici 63.
Heterozygoti H63D nejsou spojeni s přetížením železem. U homozygotů C282Y/H63D je mírné až středně závažné přetížení železem, které je klinicky manifestní až v přítomnosti precipitujících faktorů (alkoholismus, steatóza). Ostatní mutace jsou non-HFE, tyto mohou postihovat i děti.
HFE mutace – HFE gen kóduje HFE protein (příbuzný MHC glykoproteinu I), který se za normálních okolností spojuje s β2 mikroglobulinem a transferinovým receptorem 1 (TfR1) – komplex HFE – B2G – TfR1. Při mutaci C282Y je tato interakce porušena a mutantní HFE protein zůstává retinován intracelulárně s porušením resorpce železa pomocí TfR1 střevními buňkami v kryptách. To má ale za následek upregulaci DMT1 (divalent metal transporter) v kartáčovém lemu enterocytů s neúměrným zvýšením resorpce železa. Non-HFE mutace – hepcidin je peptid secernovaný v játrech, který inhibuje ferroportin a tím i transport železa na bazolaterální straně membrány a uvolnění železa z makrofágů a ostatních buněk. Hepcidin je ovlivňován signálními molekulami z jater (HFE, TfR2 a hemojuvelin). Hemochromatóza vzniká i mutací genu pro hemojuvelin, hepcidin, TfR2.

na Fe2+pomocí duodenálního cytochromu B (DcytB). Poté přechází kartáčovým epitelem duodenálních enterocytů cestou DMT1. Železo přechází přes bazolaterální membránu do krve, přes ferroportin
a ferroxidázou hefestinem (Heph). V cirkulaci se váže na plasmatický transferina je transportován na místo svého určení. Velká část Fe2+ je použita k syntéze erytrocytů v kostní dřeni a tvorbě hemoglobinu. Na konci života jsou staré erytrocyty fagocytovány makrofágy a železo je vráceno do oběhu pomocí ferroportinu. Toto vše je regulováno hepcidinem.
- 2. Sekundární přetížení železem – má stejné klinické příznaky jako hereditární hemochromatóza. Vzniká při:
- chronických poruchách erytropoézy (inefektivní erytropoéza jako sideroblastická anémie a talasémie). Při těchto poruchách je zvýšena absorpce železa, přetížení železem způsobené transfuzemi a navíc jsou pacienti často neadekvátně léčeny železem při mikrocytóze.
- porphyria cutanea tarda, kdy je přetížení železem většinou malé na to aby způsobilo postižení tkání. Někteří pacienti s PCT mají defekt HFE genu a současně hepatitidu C.
- hereditární aceruloplasminémii, poruše mobilizace železa při deficitu ceruloplasminu (ferroxidáza), což působí přetížení hepatocytů.
- 3. Zvýšený perorální příjem železa – je vzácně příčinou hemochromatózy:
- popsán v Jižní Africe, kde určité skupiny obyvatelstva konzumovaly alkoholické nápoje, které byly zkvašené v železných nádobách
- výjimečně u pacientů, kteří dlouhou dobu užívali preparáty železa perorálně, zde ovšem musela být v pozadí i určitá genetická porucha.
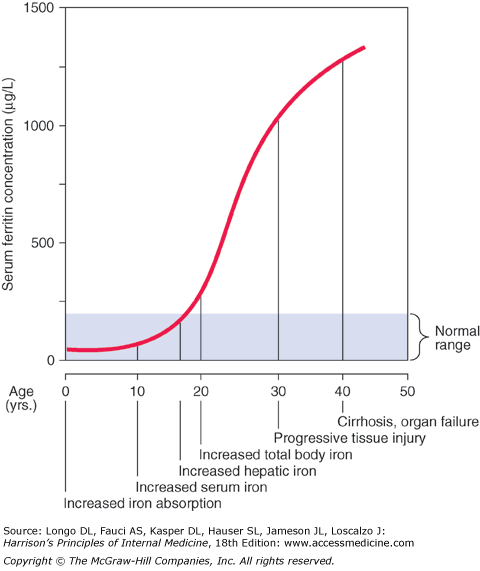
Klinický obraz – manifestace choroby je závislá na mnoha faktorech:
- protektivní – menstruační krvácení, dárcovství krve
- negativní – alkoholismus
Přetížení železem se vyvine u 30 % mužů, cirhóza u 6 % mužů, ale jen 1 % žen. Homozygoti C282Y progredují přes následující stádia:
- Genetická predispozice bez abnormalit
- Asymptomatické přetížení železem
- Časné symptomatické stádium – nespecifické příznaky (letargie, artralgie, změna barvy kůže, ztráta libida, objevení diabetes mellitus).
- Pozdní symptomatické stádium s orgánovou manifestací – známkami pokročilého onemocnění jsou hepatomegalie s projevy jaterního selhání a ikterem, hyperpigmentace, artropatie, arytmie a srdeční selhání, atrofie varlat.
1. Postižení jater – játra jsou postiženy jako první a při klinické manifestaci je hepatomegalie přítomna u > 95 % pacientů. V játrech je železo uloženo zejména ve formě feritinu a hemosiderinu. Zpočátku dochází k depozici železa v periportálních hepatocytech (zejména v lysozomech), následně se ukládá železo difuzně s aktivací makrofágů (Kupfferovy buňky) a postupným vznikem makronodulární nebo smíšené cirhózy. Játra bývají zvětšena s uzlovitou přestavbou a spolu s pankreatem mají rezavou barvu.
CAVE Hepatocelulární karcinom je nejčastější příčinou smrti (vyskytuje se u 30 % pacientů ve stádiu cirhózy). Incidence je častější u mužů, ve starším věku a vždy jen u cirhotiků.
2. Postižení kůže – v pokročilém stádiu choroby je výrazná kožní pigmentace kovové nebo břidlicové barvy, někdy označovaná jako bronzová, někdy je difuzní, často ale jen lokalizované (nejčastěji na obličeji, krku, extenzorových plochách dolního předloktí a dorzech rukou, dolní části nohou a výrazně i v jizvách). V okolí synoviálních buněk kloubů se nachází depozita železa. Toto vše koreluje s množstvím železa v játrech.

3. Diabetes mellitus – u 65 % pacientů v pokročilém stádiu choroby, častěji s rodinnou anamnézou diabetu (přímá destrukce pankreatických ostrůvků depozity železa precipituje ostatní rizikové faktory). Léčba je stejná jako u ostatních forem diabetu.
4. Artropatie – u 25 – 50 % symptomatických pacientů, nejčastěji ve věku okolo 50 let věku. Nejdříve jsou postiženy klouby ruky, zejména 2. a 3. MCP kloubu, poté může následovat postižení zápěstí, kyčlí, kolen a kotníků. Patofyziologický důvod je neznámý. I přes opakované venepunkce má artropatie tendenci k progresi. Na RTG jsou cystické změny subchondrální kosti, ztráta kloubní chrupavky, difuzní demineralizace, hypertrofická kostní proliferace a kalcifikace synovie.
5. Srdeční postižení – u 15 % pacientů v pokročilém stádiu choroby. Manifestuje se jako:
- městnavé srdeční selhání (65 % pacientů s kardiální manifestací). Morfologicky kardiomegalie, často mylně označena jako idiopatická dilatační kardiomyopatie.
- arytmie (SVT, SVES, fibrilace a flutter síní, AV blokády různého stupně)
6. Centrální hypogonadismus – vVzniká důsledkem infiltrace hypotalamu a hypofýzy se snížením produkce gonadotropinů. Má klasickou manifestaci (ztráta libida, impotence, atrofie varlat, gynekomastie, prořídnutí ochlupení a amenorea) a může být prvním příznakem choroby.
7. Ostatní – raritně se manifestuje jako adrenální insuficience, hypotyreóza a hypoparatyreóza.
Diagnostika – je nutná brzká diagnostika a léčba před trvalým poškozením orgánů. Algoritmus diagnostiky:
- 1. Anamnéza – nutná pečlivá RA, abusu alkoholu, dotaz na obsah železa ve stravě, dotaz na požití vysokých dávek vitamínu C (podporuje resorpci železa).
- 2. Vyloučení hematologické příčiny přetížení železem
- 3. Stanovení množství železa v organismu – železo v séru, vazebná kapacita železa (saturace transferinu), feritin v séru, dále biopsie jater se stanovením železa v jaterní sušině. Sérová hladina železa, saturace transferinu i feritinu jsou zvýšené již časně, ale mají nízkou senzitivitu i specifitu:
- sérová hladina železa je zvýšena i u alkoholické steatohepatitidy
- saturace transferinu ˃ 50 % by měla vést k podezření na hemochromatózu
- feritin je dobrým indikátorem celkových zásob železa v organismu. Nárůst koncentrace feritinu o 1 mg/l = nárůst celkových zásob železa o 5 mg. Koncentrace feritinu ˃ 1000 mg/l by měla vést k podezření na hemochromatózu. K falešné pozitivitě může vést přítomnost zánětu a hepatocelulární nekrózy (CAVE Feritin je protein akutní fáze).
- 4. MRI jater – jistými algoritmy lze nyní stanovit koncentraci železa v játrech.
- 5. Genetické vyšetření – indikováno při přetrvávajícím podezření.
- 6. Biopsie jater – význam při diagnostice hemochromatózy je nyní omezený pro širokou dostupnost genetického vyšetření. Jediná potvrdí přítomnost cirhózy (vždy je nutné provést barvení na železo).

Terapie
- 1. Venepunkce – ideální je odstranění 500 ml krve 1 – 2 x týdně, až do poklesu feritinu na hladinu 50 – 100 mg/l. Odstranění 1 ml krve = odstranění 0,5 mg železa. Cílem je dosažení hladiny ferritinu < 50 ug/l. Udržovací léčba se provádí v závislosti na koncentraci ferritinu; obvykle je dostatečná jedna flebotomie každé tři měsíce.
- 2. Chelatační terapie – desferrioxamin jen zcela výjimečně tam, kde nemocný netoleruje krevní odběry (hemolytické anémie, dyserytropoetické syndromy). protože neboť lék je málo účinný a velmi nákladný.
- 3. Erytrocytaferéza – pouze u nemocných neschopných dostatečné syntézy plazmatických bílkovin odebíraných při flebotomii.
- 4. Transplantace jater – indikována při end-stage fázi. Musí ji předcházet normalizace celkových zásob železa v organismu.
Prognóza – u neléčené hemochromatózy je hlavní příčinou smrti dekompenzace jaterní cirhózy, hepatocelulární karcinom, diabetes mellitus a kardiomyopatie. Nutný je pravidelný screening hepatocelulárního karcinomu (ultrazvuj kater a AFP á 6 měsících). Při včasném zahájení léčby je prognóza nemocných výborná.
Depistáž – při stanovení diagnózy hemochromatózy je nutné provést screening u rodinných příslušníků:
- u všech stanovení železa a feritinu v séru a saturace transferinu
- u příbuzných I. stupně genetické testování na přítomnost mutace C282Y a H63D
Poznámka – z praktického hlediska není nutné děti testovat před dovršením 18 let věku. Dárcovství krve odebrané nemocným s hemochromatózou je většinou odmítáno.