Epidemiologie – roční incidence 1:25 000 / rok, 1: 6500 / rok u osob starších 65 let, průměrný věk manifestace 67 let.
Etiologie – u většiny pacientů je příčina neznáma, zajímavosti je že 5 – 6 % zdravých jedinců > 70 let obsahuje potenciálně premaligní mutace které jsou spojené s klonální expanzi. Pravděpodobné predisponující faktory:
- zevní prostředí – ionizující záření, některé viry, alkylační cytostatika (AML po 4 – 6 letech), inhibitory topoizomerázy II (AML po 1 – 3 letech).
- vrozené – Downův syndrom, Klinefelterův syndrom, Fanconiho syndrom, Wiskottův – Aldrichův syndrom, Kostmanův syndrom a jiné.
Patogeneze – leukemické kmenové buňky vznikají maligním zvratem normálních hemopoetických kmenových buněk. Jejich následným dělením vzniká populace nezralých leukemických blastů. Jelikož jsou nesmrtelné, mají dostatek času na to, aby akumulovaly nezbytné mutace, které vedou ve svém důsledku k vyvázání z homeostázy organismu;
- mutace genu pro receptorovou tyrosinkinázu – nejčastěji chromozomální translokace, – prototypem jsou myeloproliferativní onemocnění – např. CML: translokace t(9,22), během níž dochází k přesunu genu pro receptorové tyrosinkinázy ABL do oblasti BCR. Vzniká chimérícký fúzní gen bcr-abl s tyrosinkinázovou aktivitou. Zatímco receptorová tyrosikinázová abl se aktivuje pouze vazbou specifického ligandu, její translokovaná forma bcr-abl je aktivovaná kontinuálně. Následkem toho signalizuje i kontinuálně do jádra proliferativní stimul, který působí zvýšenou autonomní proliferaci klonu.
- mutace genů pro hematopoetické transkripční faktory – působí inhibici hematopoetického transkripčního faktoru, to má za následek diferenciační blok, což se projeví přítomností určitého procenta nezralých buněk (leukemických blastů) v kostní dřeni popř. i periferní krvi. Vzniká tak autonomní klon s dysplastickými rysy, který ale zatím nemá potřebnou proliferativní aktivitu. Prototypem je myelodysplastický syndrom.
Dojde – li ke kombinaci obou těchto mutací dochází k transformaci z chronické fáze choroby do akutní leukémie. V krevním obraze je patrná leukocytóza s přítomným hiatus leucaemicus – přítomny nezralé dysplastické formy vedle normálních zralých buněk, které ubývají díky agresivní expanzi leukemického klonu. Generační poločas leukemické populace je ve srovnání s normálními buňkami delší (více než 60 hodin), podobně je i delší zdvojovací čas. Normální kmenové buňky jsou leukemickou populací postupně utlačeny, díky čemuž vzniká anémie a trombocytopenie.
Klasifikace – nyní se používá klasifikace WHO z roku 2016, ve které je patrný výrazný pokrok na poli cytogenetiky. Obecně lze říct, že AML se dělí na následující podskupiny:
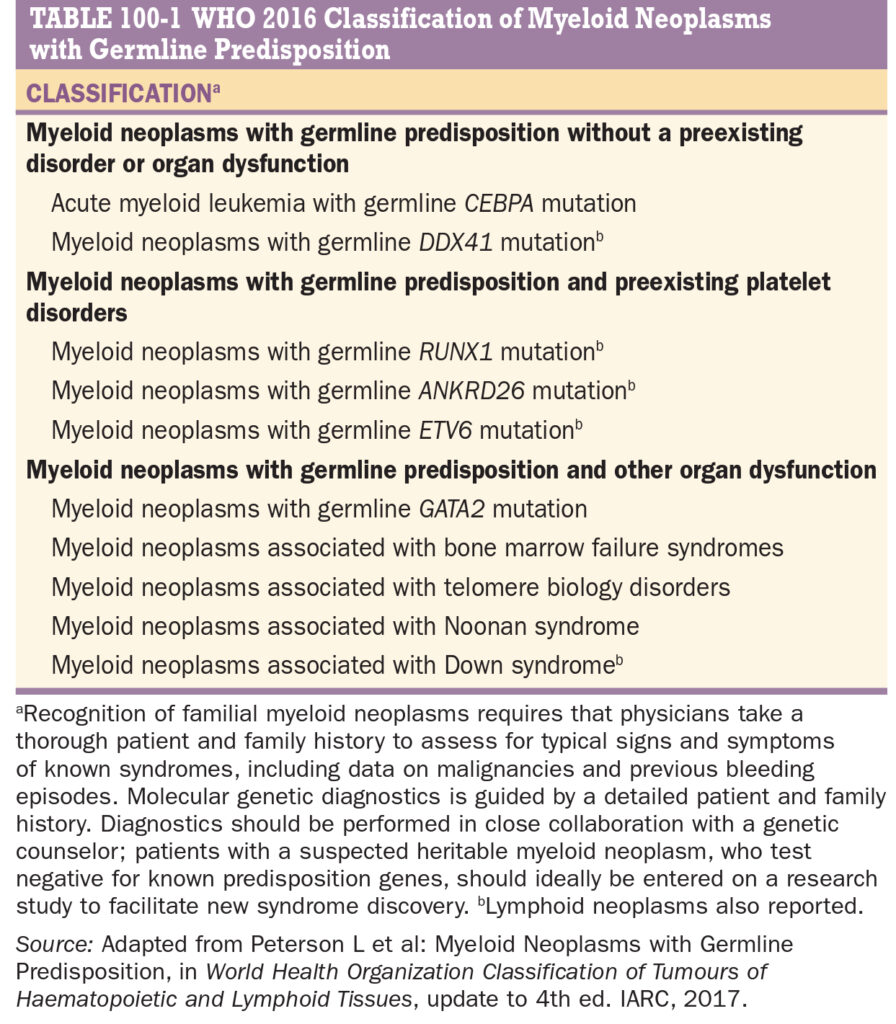
- AML s vrozenými predispozicemi (viz tabulka)
- AML s rekurentními genetickými abnormalitami
- akutní promyelocytární leukémie s PML-RARA
- související s MDS
- související s Downovým syndromem
- nespecifikované (zde podobné jako při FAB)
- myeloidní sarkom

Klinický obraz – příznaky jsou nespecifické a značně pestré (chřipkovité příznaky, únava, slabost, subfebrilie, pocení), u 50 % pacientů je historie příznaků kratší než 3 měsíce. Častá je abnormální krvácivost (petechie, hematomy, epistaxe, krvácení z dásní). Těžké krvácení je vzácné, může provázet akutní promyelocytární leukémii. Objektivně lze nalézt bledost (anémie), krvácivé projevy (sítnice, CNS), může být mírná hepatosplenomegalie, bolesti kostí. Pro monocytární formy jsou typické infiltraci kůže, dásní, měkkých tkání nebo mening.

{kind=link}
- Krevní obraz – typicky je přítomna leukocytóza, ale může být i leukopenie, je přítomno různé procento blastů, při absenci středních vývojových řad (promyelocyty, myelocyty, metamyelocyty) – hiatus leucaemicus.
- Biopsie kostní dřeně – morfologické vyšetření umožňuje stanovení základních typů akutní leukémie, ovšem již ne rozlišení subtypů. Většinou je základním kritériem počet blastů v kostní dřeni > 20 %.
- Imunofenotypizace průtokovou cytometrií – orientační stanovení typu leukémie podle CD subtypů.
| CD19 | CD7 | CD33 | CD13 | Ia | TdT | |
| Myeloidní linie | – | – | + | + | + | – |
| Lymfoidní linie B | + | – | – | – | + | + |
| Lymfoidní linie T | – | + | – | – | – | + |
- Pozitivita reakce na myeloperoxidázu.
- Screening mutací a přestaveb genů.
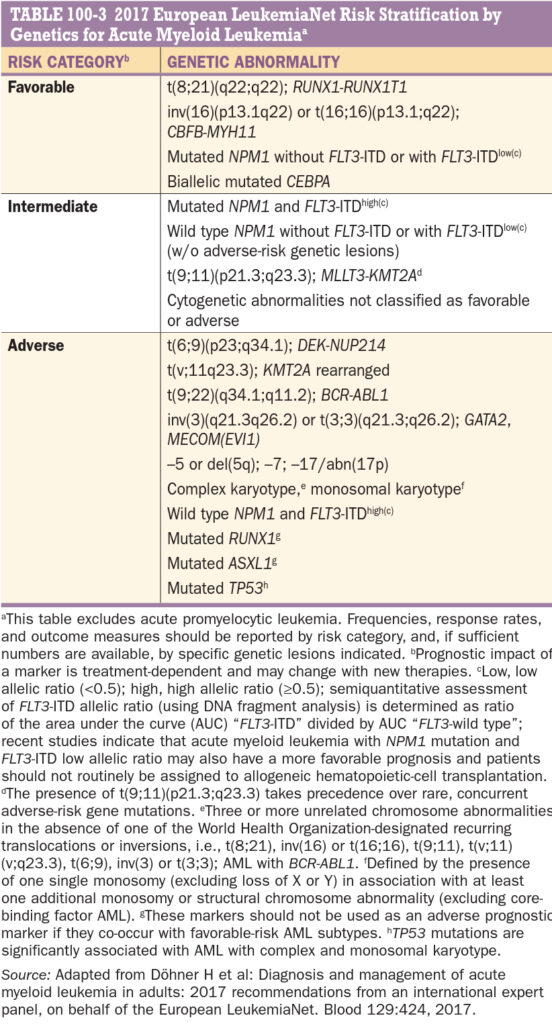
Prognostická kritéria podle typu
- příznivá – t(8,21), inv(16), t(16,16), t(15,17)
- intermediární – normální karyotypy nebo nezařazené
- velmi nepříznivá – del(7), del(5), t(6,11), t(9,11), t(11,19), t(11,17)
- Mezi nepříznivé prognostické faktory patří nález genu pro mnohočetnou lékovou rezistenci (MDR-1), hyperleukocytóza, nedosažení kompletní remise indukční léčbou.
Terapie – prvním úkolem je zhodnocení stavu nemocného a určení intenzity jeho léčby. Obecně lze říct, že většina nemocných mladších 60 – 65 let je lečena intenzivně.
Intenzivní léčebný přístup:
- Indukční léčba – režim 3 + 7 (3 dny antracyklinů + 7 dnů cytarabinu). Event. na základě podskupin lze přidat midostaurin/gemtuzumab ozogamycin/CPX-351. Následuje vyšetření kostní dřeně s následující úpravou terapie dle výsledku a poté postremisní chemoterapie.
- Konsolidační léčba – většinou vyššími dávkami cytarabinu (HiDAC).
- Pokud je riziko relapsu bez provedení alogenní transplantace kostní dřeně – HSCT (dáno vstupním molekulárně – cytogenetickým vyšetřením) vyšší než 35 – 40 %, je alogenní HSCT indikována. Jinak řečeno, čím vyšší riziko je přítomno, tím více je alogenní HSCT indikována. Vždy by měla být provedena co nejdříve po dosažení kompletní remise. K udržení remise je potřeba konsolidační chemoterapie. Myeloablační režim je indikován pro mladší nemocné, pro starší bývá nutné redukovat intenzitu.
- U rezistentních forem bývá indikována záchranná léčba, často formou klinických studií.
Neintenzivní léčebný přístup – jelikož platí že čím starší pacient, tím horší je prognóza, měla by být všem těmto pacientům nabídnuta účast v klinických studiích. Do neintenzivních léčebných režimu patří nízkodávkovaný cytarabin, azacytidin nebo decitabin.
Další zásady:
- U každého pacienta je indikována intenzivní podpůrná léčba (antibakteriální, antimykotická, profylaxe pneumocystové pneumonie cotrimoxazolem).
- V případě hyperleukocytózy je indikována podání dexametazonu, nízkodávkované chemoterapie, v případě leukostázy doplněné o leukaferézu.
- V případě postižení CNS je indikováno intrathékální podání cytarabinu, doplněné o dexametazonu i.v. k prevenci arachnoiditidy.
Blíže k terapii viz. „Červená kniha“ dostupná online zde: https://www.hematology.cz/doporuceni/klinika-files/Doporuceni_CHS_CLS_JEP-Cervena_kniha.pdf
Prognóza – neléčená je během několika měsíců smrtelná. Prognóza záleží na typu.
Promyelocytární leukémie
Epidemiologie – podstatou PML je klonální expanze patologických krvetvorných buněk, jejíchž zrání se zastavilo na úrovni promyelocytu. Tvoří cca 8 % všech AML, narozdíl od nich se vyskytuje v mladším věku (medián 45 let), není rozdíl v četnosti mezi pohlavími.
Patofyziologie – podstatou choroby je genetická mutace, v 97 % případů translokace genů pro receptor kyseliny alfa-retinové (RARα) z chromozómu 17 na chromozóm 15 do oblasti označované PML – t(15;17). Vzácně jsou odpovědné i jiné, tzv. variantní translokace. Všechny mutace vedou ke změně struktury receptoru pro kyselinu retinovou. Za normálních okolností vede i malé množství kyseliny alfa-retinové vázané na tento receptor k indukci diferenciace buněk. Při zmiňované mutaci tato fyziologická hladina nevede k pokračování diferenciace s jejím zastavením na úrovni promyelocytu. Pokračování diferenciace je umožněno až vysokou hladinou kyseliny retinové podávané terapeuticky.
Klinický obraz – hlavním a nejnebezpečnějším příznakem PML je krvácení v jakékoliv lokalizaci, nejčastější kožní a slizniční. Nejzávažnější formou je krvácení do CNS, která často vede ke smrti nemocného. Závažné formy krvácení se objevují často během několika dnů – PML je proto považována za hematologickou emergenci. Příčinou krvácení je trombocytopenie a zejména koagulopatie (spotřebováním koagulačních faktorů při masivním vyplavování prokoagulačních substancí z leukemických buněk a také excesivní fibrinolýzou). Obraz se tak podobá rozvinuté diseminované intravaskulární koagulaci. Mimo krvácení se PML může projevovat příznaky anémie a neutropenie (infekce).
Diagnostika – v krevním obraze je typicky pancytopenie. Případná leukocytóza patří spolu s trombocytopenií k rizikovým faktorům krvácení do CNS.
CAVE Leukemické promyelocyty se nemusí vyplavovat do periferní krve, proto je ke stanovení diagnózy zásadní až vyšetření kostní dřeně (nezřídka hypocelulární a špatně aspirovatelná).
U většiny pacientů je přítomna koagulopatie, zejména snížená hladina fibrinogenu, často prodloužení PT a aPTT. Není specifické, přesto:
U každého nemocného s koagulopatií a abnormálním krevním obrazem budícím podezření na akutní leukémii je nutné na tuto diagnózu pomyslet.
Vyšetření kostní dřeně – přítomny atypické promyelocyty s hypergranulacemi, přítomny bývají i Aureovy tyče (azurofilní krystalizovaná granula), které jsou často tak četné, že připomínají otýpky („faggot cells„)
Průtoková cytometrie obvykle prokáže pozitivitu myeloperoxidázy, CD13, CD33 a negativitu HLA-DR. Zásadním k průkazu PML je cytogenetické vyšetření (FISH) a molekulárně genetické vyšetření (PCR), které prokáže patologickou translokaci.
Mezi faktory zhoršující prognózu patří leukocytóza, vyšší věk a některé typy mutací.
Diagnóza = morfologický nález patologických promyelocytů v kostní dřeni + průkaz PML/RARα popř. t(15;17),
Terapie – v prvních dnech až dvou týdnech je zásadní zvládnutí syndromu DIC a závažného krvácení (trombocyty udržovat > 50, čerstvá mražená plazma, antitrombin, fibrinogen udržovat > 1,5 g/l). Podobně je v tomto období zcela nevhodné provádět jakákoliv invazivní vyšetření popř. zavádět centrální žilní katetr, pokud k tomu nemáme zcela zásadní důvod.
Vždy je nutné zahájit podávání kyseliny all-transretinové (ATRA) co nejdříve po vyslovení podezření na APL a bez čekání na potvrzení diagnózy.
V léčbě první linie s nízkým rizikem se používá ATRA v kombinaci s oxidem arsenitým. Při vysokém riziku chemoterapie (nejčastěji antracykliny) v kombinaci s oxidem arsenitým, ATRA popř. s gemtuzumabem ozogamicinem. I zde jsou fáze indukce a konsolidace a zcela zásadní je udržovací léčba, při které se používá ATRA, 6-merkaptopurin a metotrexát.
Hlavní komplikací léčby je diferenciační syndrom, k terý vzniká u cca 20 % nemocných léčených ATRA nebo oxidem arsenitým v indukční fázi terapie. Rozvíjí se obvykle během několika dnů po zahájení léčby spolu s nárůstem leukocytózy a je pravděpodobně způsoben uvolněním zánětlivých cytokinů s mohutně dozrávajících promyelocytů. Pokud je plně vyjádřen, projevuje se nárůstem hmotnosti s otoky, pleurálními a perikardiálními výpotky, horečkou, dušností, bolestmi hlavy, intersticiálními plicními infiltráty, v těžkých případech také hypotenzí, multiorgánovým selháním až smrtí pacienta (pro zapamatování a velmi odlehčeně připomíná kombinaci srdeční selhání + chřipka). Léčba spočívá v podávání vysokých dávek dexametazonu při prvních příznacích diferenciačního syndromu, u těžkých forem je nutné na přechodnou dobu přerušit léčbu pomocí ATRA nebo oxidu arsenitého.
CAVE Na diferenciační syndrom je potřeba pomyslet při nárůstu hmotnosti s otoky a pleurálním výpotkem 2-5 dnů po zahájení léčby.
CAVE U oxidu arsenitého je také potřeba pohlídat délku QT intervalu. Při jeho prodloužení nad 500 milisekund je doporučeno podávání ATO přerušit (do opětovného zkrácení pod 460 ms). Samozřejmostí je podávání magnézia a pokud možno vysazení ostatních léků, které vedou k prodloužení QT intervalu.