Úvod
Prvním příznakem je nejčastěji progredující dušnost se suchým, dráždivým kašlem. Později se objevuje hemoptýza, pískoty, bolest na hrudi a opacity na RTG S+P. Existuje více než 200 nosologických jednotek (může být pouze izolované poškození plic nebo plicní poškození může být součástí celkové choroby, nejčastěji systémové choroby pojiva).
Klasifikace
| Plicní choroby se zánětem a fibrózou | |
| Ze známé příčiny | |
| Azbestóza | |
| Inhalace plynů a výparů | |
| Léky | nitrofurantoin |
| amiodaron | |
| zlato | |
| Radiační poškození | |
| Aspirační pneumonie | |
| Reziduum po ARDS | |
| Poškození spojené s kouřením | Deskvamující intersticiální pneumonie |
| Respirační bronchiolotida | |
| Granulomatóza z Langerhansových buněk | |
| Z neznámé příčiny | |
| Idiopatické intersticiální pneumonie | Idiopatická plicní fibróza |
| Akutní intersticiální pneumonie | |
| Bronchiolitis obliterans s organizující pneumonií | |
| Nespecifická intersticiální pneumonie | |
| Systémové choroby pojiva | Systémový lupus erythematosus |
| Revmatoidní artritida | |
| Ankylozující spondylitida | |
| Systémová skleróza | |
| Sjögrenův syndrom | |
| Polymyozitida / dermatomyozitida | |
| Syndromy s plicním krvácením | Goodpastureův syndrom |
| Idiopatická plicní hemosideróza | |
| Izolovaná plicní kapilaritida | |
| Plicní alveolární proteinóza | |
| Plicní choroba s infiltrací lymfocyty | většinou asociované se systémovými chorobami pojiva |
| Eozinofilní pneumonie | |
| Lymfangioleiomyomatóza | |
| Amyloidóza | |
| Genetické poruchy | Tuberózní skleróza |
| Neurofibromatóza | |
| Niemann-Pickova choroba | |
| Gaucherova choroba | |
| Syndrom Heřmanského – Pudláka | |
| Plicní choroby asociované s postižením GIT | Crohnova choroba |
| Ulcerózní kolitida | |
| Primární biliární cirhóza | |
| Chronická aktivní hepatitida | |
| Graft-versus-host disease | transplantace kostní dřeně |
| transplantace orgánů | |
| Granulomatozní plicní choroby | |
| Ze známé příčiny | |
| Hypersenzitivní pneumonie | expozice organickému prachu |
| Anorganický prach | berilióza, silikóza |
| Z neznámé příčiny | |
| Sarkoidóza | |
| Granulomatózní vaskulitida | |
| Granulomatóza s polyangitidou | |
| Syndrom Churg-Straussové | |
| Bronchocentrická granulomatóza | |
| Lymfomatoidní granulomatóza |
Dělí se na dvě základní skupiny:
- Granulomatózní plicní choroby – dochází k akumulaci T lymfocytů, makrofágů a epiteloidních buněk, které se organizují do jednotlivých útvarů (granulomů). Tyto choroby mohou progredovat až do plicní fibrózy nebo plicní poškození může být zanedbatelné. Diff. dg. je nutné odlišení od sarkoidózy a hypersenzitivní pneumonitidy.
- Plicní choroby se zánětem a fibrózou – vyvolávajícím faktorem je poškození epitelu, které vyvolá zánět alveolů. Pokud zánět přejde do chronicity, zasahuje i intersticium a cévy s následnou progresí do plicní fibrózy, která postihuje ventilační i oxygenační funkce.
Patofyziologie
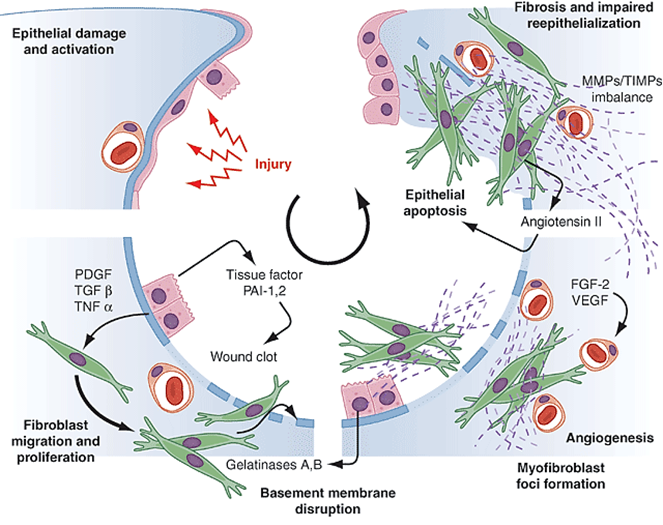
FGF-2 fibroblast growth factor 2, MMP metaloproteinázy, PAI2 inhibitoru aktivátoru plazminogenu, PDGF platelet derived growth factor, TNF-α tumor necrosis factor α, TIMP – tkáňový inhibitor mataloproteináz, TGF-β transforming growth factor β, VEGF vascular endotelial growth factor
Diagnostika
Trvání choroby
Akutní průběh (dny až týdny) – pro obraz difuzních opacit na rentgenu srdce a plic může být záměna za atypické pneumonie, alergie (léky, mykotické a helmintické infekce), akutní intersticiální pneumonie, eozinofilní pneumonie, hypersenzitivní pneumonitida.
Subakutní průběh (týdny až měsíce) – časně se příznaky mohou objevit u všech intersticiálních plicních procesů, ale typické je pro sarkoidózu, polékové syndromy s plicním krvácením, bronchiolitis obliterans s organizující pneumonií, pneumonitidu asociovanou se systémovým lupus erythematosus.
Chronický průběh (měsíce až roky) – chronický průběh je typický pro většinu intersticiálních plicních procesů např. idiopatická plicní fibróza, granulomatóza z Langerhansových buněk, pneumokoniózy a postižení plic při systémových chorobách pojiva.
Epizodický průběh – je neobvyklý, např.:eozinofilní pneumonie, hypersenzitivní pneumonie, bronchiolitis obliterans s organizující pneumonií, vaskulitidy, syndromy s plicním krvácením, syndrom Churg – Straussové.
Věk
20 – 40 roků – sarkoidóza, postižení plic při systémových chorobách pojiva, lymfangioleimyomatóza, granulomatóza z Langerhansových buněk, geneticky podmíněné nemoci (familiární idiopatická plicní fibróza, Gaucherova choroba, syndrom Heřmanského-Pudláka)
Více než 60 let – idiopatická plicní fibróza
Pohlaví
Premenopauzální ženy – lymfangioleimyomatóza, plicní postižení u tuberózní sklerózy
Ženy – syndrom Heřmanského-Pudláka, postižení plic při systémových chorobách pojiva (s výjimkou plicního procesu u revmatoidní artritidy, který je častější u mužů)
Muži – idiopatická plicní fibróza, plicní proces u revmatoidní artritidy, vzhledem k častější profesní zátěži i pneumokoniózy
Rodinná anamnéza
Familiární plicní fibróza (rizikovým faktorem je kuřáctví, starší věk a mužské pohlaví), neurofibromatóza, tuberózní skleróza (AD dědičnost), sarkoidóza (častější výskyt při pozitivní rodinné anamnéze).
Anamnéza kouření
U IPF a familiární plicní fibrózy 70 % pacientů kouří cigarety. Granulomatóza z Langerhansových buněk, deskvamující intersticiální pneumonie, respirační bronchioloitida, Goodpastureův syndrom, plicní alveolární proteinóza jsou většinou spojeny s kuřáctvím.
Profesní a enviromentální vlivy
Nutné odebrání podobné profesionální anamnézy (farmářská plíce) a seznámit se i s koníčky pacienta (hypersenzitiovní pneumonitis). Obtíže mohou vymizet jen několik dnů od zanechání expozice alergenu a naopak po reexpozici rychle recidivují.
Ostatní důležité údaje anamnézy
Cestovatelská anamnéza důležitá u helmintické infekce (často působí plicní eozinofilii), rizikové chování pro HIV (během HIV infekce se mohou přidat i organizující se pneumonie, lymfocytární intersticiální pneumonitida a difuzní alveolární hemoragie).
Příznaky vycházející z dýchacího ústrojí
Dušnost – v popředí zejména u idiopatické intersticiální pneumonie, hypersenzitivní pneumonitidy, bronchiolitis obliterans s organizující pneumonií, sarkoidózy, eoziofilní pneumonie a granulomatózy z Langerhansových buněk
Výrazný nález na rentgenu srdce a plic bez dušnosti na počátku chororby – sarkoidóza, silikóza, granulomatóza z Langerhansových buněk, hypersenzitivní pneumonitida, lipoidní pneumonie
Bronchitické fenomény – vzácné u intersticiálních procesů, ale popsány u chronické eozinofilní pneumonie, syndromu Churg – Straussové, respirační bronchiolitidy, sarkoidózy
Dyskomfort na hrudníku – netypický, popisována u sarkoidózy
Spontánní pneumothorax – může u granulomatózy z Langerhansových buněk, tuberózní sklerózy, lymfangioleiomyomatózy, neurofibromatózy
Hemoptýza – netypická, může u syndromů s difuzním alveolárním krvácením, tuberózní sklerózy, lymfangioleiomyomatózy, granulomatózních vaskulitid
Slabost, váhový úbytek – typicky u všech intersticiálních plicních procesů
Fyzikální vyšetření
Nálezy jsou většinou nespecifické. Nejčastější nálezy jsou tachypnoe, endinspirační suché krepitace bilaterálně nad bázemi (u většiny zánětlivých, vzácně u granulomatózních zánětlivých intersticiálních procesů, mohou být přítomny i při absenci RTG změn), endinspirační vrzoty a pískoty (časté u bronchiolitidy). V pozdních stádiích intersticiálních procesů známky plicní hypertenze a cor pulmonale, cyanóza a paličkovité prsty.
Laboratorní nález
Imunologické vyšetření – ANA, revmatoidní faktor (často nespecifický pozitivní), ANCA, anti GBM (při podezření na vaskulitidy).
Biochemické vyšetření – LD – často nespecificky pozitivní, sACE – u sarkoidózy
EKG – normální nález, dokud se nevyskytne plicní hypertenze (pravotyp osy, známky hypertrofie pravé síně a komory), lze potvrdit echem
Zobrazovací metody
RTG srdce a plic – časně bilaterálně bazálně retikulární kresba nebo opacity, odpovídající infiltraci alveolů, většinou není nález specifický pro určitou chorobu. Některé jednotky postihují predilekčně horní plicní pole (sarkoidóza, granulomatóza z Langerhansových buněk, chronická hypersenzitivní pneumonitida, silikóza, berylióza, plicní forma revmatoidní artritidy, ankylozující spondylitida). Ve většině případů závažnost rentgenologických změn nemusí odpovídat klinické závažnosti.
CT – metodou volby je HRCT (high-resolution CT), který odhalí intersticiální změny i u pacientů s normálním rentgenem srdce a plic. Je přínosné i k odhalení jiné koexistujicí choroby (adenopatie mediastina, tumoru, emfyzému apod.) a event. zaměření místa plicní biopsie.
Funkční vyšetření plic
Spirometrie – restriktivní typ křivky, ↓TLC, FRC, RV. FEV1 je také snížené, ale ne obstrukcí, ale díky ↓TLC → FEV1/FVC je normální.
Difuzní kapacita – ↓DLCO – vysoká senzitivita, nízká specificita, není korelace s pokročilostí procesu. Příčinou je ztluštění a redukce respirační plochy a zhoršení poměru ventilace a perfúze (V/Q).
Krevní plyny ↓pO2 – hypoxémie je vlivem poruchy V/Q a projevuje se zejména při zátěži, ↑pCO2 – většinou známkou end-stage disease.
Při zátěžovém funkčním vyšetření dochází k neadekvátní tachypnoi s poklesem pO2 i u pacientů s normální klidovou hladinou pO2 a vzestupu poměru VD/VT (mrtvý objem / dechový objem), protože nedochází k adekvátnímu vzestupu VT (dechového objemu). Ke zhodnocení celkové kondice je vhodné provedení 6 minutového testů chůze.
Bronchoskopické vyšetření s bronchoalveolární laváží
Bronchoalveolární laváž – může být užitečná k diagnostice sarkoidózy, hypersenzitivní pneumonitidy, syndromů difuzní alveolární hemoragie, tumorů, plicní alveolární proteinózy. Interpretace:
| Choroba | Nález v BAL |
| Sarkoidóza | lymfocytóza, charakteristicky CD4:CD8 ˃ 3,5 |
| Hypersenzitivní pneumonitida | výrazná lymfocytóza (˃ 50 %) |
| Bronchiolitis obliterans s organizující pneumonií | pěnové makrofágy, CD4:CD8 ˃ snížené, smíšená buněčnost |
| Eozinofilní plicní choroba | výrazná eozinofilie (> 25 %) |
| Difuzní alveolární krvácení | siderofágy, erytrocyty |
| Intersticiální poškození indukované léky | atypická hyperplazie pneumocytů II. typu |
| Oportunní infekce | Pneumocystis jiroveci, mykotické změny, CMV transformované buňky |
| Tumory | maligní buňky |
| Alveolární proteinóza | mléčně zbarvená lavážní tekutina, pěnové makrofágy, lipoproteinový intraalveolární materiál |
| Lipoidní pneumonie | makrofágy s fagocytovanými lipidy |
| Granulomatóza z Langerhansových buněk | nadbytek CD1+ Langerhansových buněk, v elektronmikroskopicky patrna Birbeckova granula v makrofázích |
| Azbestóza a příbuzné choroby | prachové částice, částice železa |
| Berylióza | pozitivní test na berylium v lymfocytech |
| Silikóza | prachové částice v polarizovaném mikroskopu |
| Lipoidóza | akumulace specifického lipopigmentu v alveolárních makrofázích |
Biopsie – bronchoskopicky s odběrem 4 – 8 bioptických vzorků. Je metodou volby ke konfirmaci diagnózy a měla by být provedena před zahájením léčby (zejména při sarkoidóze, lymfangitické karcinomatóze, eozinofilní pneumonii, Goodpastureovu syndromu). Pokud není vyšetření průkazné je indikována VATS nebo torakotomie s odběrem materiálu, kdy by měl být proveden odběr minimálně ze dvou laloků (kontraindikací je pokročilá kardiovaskulární choroba, kaverny, end-stage disease, stáří).
Terapie
Choroby jsou většinou k léčbě rezistentní a často progredují. Fibrotizace je již ireverzibilní, proto je hlavním cílem terapie zpomalení progrese choroby (zánětu).
- Farmakoterapie – glukokortikoidy jsou základem terapie, protože působí potlačení alveolitidy. Indikovány u eozinofilní pneumonie, kryptogenní organizující pneumonie, intersticiálních chorob spojených se systémovými chorobami pojiva, sarkoidózy, hypersenzitivní pneumonitidy, akutní expozice anorganickým prachům, akutní radiační pneumonitidy, syndromu difuzních alveolární hemoragie a polékových intersticiálních chorob, při expozici organickým prachům jsou indikovány jak v akutním, tak chronickém stádiu. Optimální dávka není stanovena, obvykle: 0,5 – 1,0 mg/kg prednisonu po dobu jednoho až tří měsíců, při stabilizaci nebo zlepšení stavu redukce na dávku 0,25 – 0,5 mg/kg po dobu dalších jednoho až tří měsíců, poté se podává udržovací dávka 0,25 mg/kg. Při příliš rychlém vysazení hrozí relaps. Při kortikorezistenci lze přidat cyklofosfamid, azathioprin (1 – 2 mg/kg). Na účinek je nutno počkat minimálně dva až tři měsíce. Při další rezistenci lze podat metotrexát, kolchicin, penicilamin a cyklosporin jako rescue terapii s nejistým účinkem.
- Léčba komplikací – léčba cor pulmonale
- Dechová rehabilitace
- Transplantace plic
1. Idiopatická plicní fibróza
Definice – idiopatická plicní fibróza (IPF) je specifická forma chronické progredující fibrotizující intersticiální pneumonitidy nejasné etiologie, objevující se primárně u dospělých jedinců, postihující pouze plíce. Nutné je vyloučení ostatních příčin plicní firbrózy.
Epidemiologie – incidence v ČR maximálně 1 : 100 tisíc (celosvětově světově cca 5/100000), věk většínou > 60 let, není rasová, etnická ani geografická prevalence.
Etiologie – neznámá. Jako suspektní vyvolávající faktor bylo identifikováno kouření cigaret, expozice prachům obsahujícím ocel, mosaz, olovo, částice prachu při řezání či leštění kamenů a expozice organickým živočišným a rostlinným antigenům a také prachu z borovicového dřeva (otázkou je, jestli popisované případy opravdu byly IPF či obraz exogenní alergické alveolitidy či pneumokoniózy). Vliv má také řada virů (u pacientů s IPF byla zjištěna vyšší incidence EBV, chřipkového viru, CMV, viru hepatitidy C a HHV-7,8, parainfluenza, HIV-1, virus spalniček, herpesvirus 6 a Mycoplasma spp). Několik studií vyjádřilo podezření i na roli GERD (chronická mikroaspirace).
Klinický obraz – projevuje se námahovou dušností se suchým kašlem, v pozdějších stádiích cyanózou event. paličkovými prsty. Poslechově bazálně je slyšitelný krepitus. Progrese je plíživá a pomalu progredující, u některých pacientů se objeví epizody akutní exacerbace (náhlé klinické a funkční zhoršení s RTG obrazem mléčného skla svědčící pro alveolitidu).
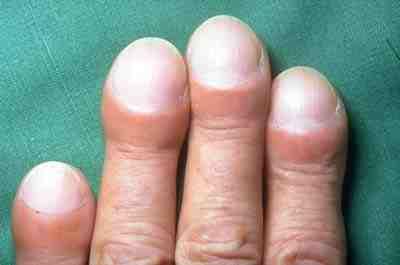
Na IPF je nutné pomyslet v případě nově diagnostikovaného intersticiálního plicního procesu nejasného původu, u pacienta typicky staršího 60 let s poslechovým nálezem inspiračních
chrůpků bilaterálně bazálně, s radiologickým obrazem plicní fibrózy oboustranně.
Diagnostika
RTG S+P a HRCT – typická je přítomnost retikulárních opacit, obvykle spojených s trakčními bronchiektaziemi, často je obraz voštiny. Zásadním je HRCT obraz UIP: časově heterogenní vzhled s obrazem ložisek fibrózy a voštiny střídajícím obraz méně postižené nebo normální tkáně. Změny obvykle postihují subpleurální oblasti a paraseptální parenchym. Zánět je obvykle minimální.


Funkční vyšetření plic – většinou restriktivní ventilační porucha, porucha difúze (redukce DlCO), hypoxémie (snížení paO2), zejména při zátěži.
Histologické vyšetření CAVE nález není difúzní, ale spíše heterogenní a střídá se s okrsky zdravé tkáně, proto není často průkazná transbronchiální biopsie a je nutné provedení chirurgické biopsie). Nejčastěji jsou postiženy bazální a periferní oblasti a rozsah histologických změn odpovídá pokročilosti choroby. Charakteristické jsou okrsky intersticiálního zánětu s lymfoplazmocytárním infiltrátem stěn alveolů a následnou hyperplazií pneumocytů 2. typu, ložiska proliferujících fibroblastů s denzní kolagenofibrózou, kavernózní změny, což jsou cysty, často ohraničené bronchiálním epitelem a vyplněné mucinem, často hyperplazie hladkého svalstva. Diferenciálně diagnosticky je podobný nález běžný při pokročilé formě pneumokoniózy (např. azbestóza), radiačního poškození, polékových změn (např. nitrofurantoin), chronické aspirace, sarkoidóze, chronické hypersenzitivní pneumonitidy, organizující eozinofilní pneumonie a hyperplazii Langerhansových buněk).
Dle 3. aktualizace doporučených postupu CPFS z 2019 musí být splněna následující kritéria:
- Vyloučení jiných příčin intersticiálních plicních procesů (domácí a profesní expozice, systémové nemoci pojiva, léková toxicita).
- Přítomnost HRCT vzorce UIP u pacientů bez plicní biopsie.
- Specifické kombinace HRCT a histopatologického UIP vzorce u pacientů s plicní biopsií.
Komplikace
- Emfyzém – dále zhoršuje prognózu
- Srdeční – na ICHS a srdeční selhání umírá 30 % pacientů
- Akutní exacerbace se projevuje akutním zhoršením dušnosti během několika dnů až jednoho měsíce, na HRCT jsou na pozadí chronických změn patrny nové okrsky mléčného skla (alveolitida). Během své choroby exacerbaci prodělá 10 – 57 % všech pacientů, tomuto odpovídá histologický obraz difuzního alveolárního poškození. Zatím nebyla nalezena žádná efektivní terapie akutních exacerbací. Často je potřeba umělá plicní ventilace, ale ani tato nebývá úspěšná s nemocniční mortalitou více než 75 % pacientů. U pacientů, kteří přežijí, dochází často k recidivě exacerbace, která již většinou končí smrtí.
Terapie
1. Farmakoterapie – základem je antifibrotická léčba:
Pirfenidon inhibuje PDGF (platelet-derived growth factor) a TGF-β. Snižuje rychlost poklesu plicních funkcí a prodlužuje vzdálenost v 6 minutovém testu chůzí. Mezi NÚ patří: nauzea a nechutenství, elevace transamináz a fotosenzitivita.
Nintenatib inhibuje PDGF, FGF (fibroblast growth factor) a VEGF (vascular endothelial growth factor). Mezi NÚ patří: průjmy a elevace transamináz. U obou léků je problematická úhrada (viz guidelines).
U pacientů, kteří nedosáhnou na antifibrotickou léčbu je doporučeno podávat N-acetylcystein v dávce 1,8 g denně (při zjištění polymorfismu TOLLIP). U symptomatického GERD indikovány PPI (preferenčně esomeprazol – jako jediný neinterferuje s pirfenidonem).
2. Nefarmakologická léčba
- UPV by u respirační insuficience způsobené IPF neměla být volena paušálně, protože mortalita při UPV je 96 % → profitují pouze pacienti, kteří mohou být transplantování z ventilátoru → UPV by měla být indikována pouze u pacientů na waiting listu na Tx plic nebo těsně před jeho zařazením.
- Komplexní rehabilitace by měla být indikována téměř ve všech případech.
Terapie akutní exacerbace IPF
- Indikována léčba na JIP.
- Pokud je pacient schopen BAL je tato vhodná.
- Nutná oxygenoterapie, vždy je vhodné upřednostnit neinvazivní ventilaci před UPV (viz výše).
- Indikované vysoké dávky kortikoidů parenterálně (první 3 dny 1 gram methylprednisolonu denně, poté v dávce 1 mg/kg).
- Při podezření na infekci ATB (doporučeny cefalosporiny III. generace, chinolony (pokrytí legionel, mykoplasmat a chlamydií), TMP/SMX k prevenci pneumocysty a antimykotika – preventivně itrakonazol. Vhodné je řídit se při indikaci antimikrobiální léčby hodnotou CD4 + T lymfocytů; pokud jejich absolutní počet klesne pod 200 buněk/µl, je nutné vykrýt preventivně pneumocystovou a mykotickou infekci vždy).
- Substituce IVIG při poklesu IgG < 6 g/l.
- Pokud je to možné a pacient je schopen je vhodná urgentní transplantace.
Prognóza – špatná (medián přežití nepřesahuje 3 roky od stanovení diagnózy), je zároveň vyšší riziko bronchogenního karcinomu.
2. Nespecifická intersticiální pneumonie
Nespecifické intersticiální pneumonie (NIP) je skupina intersticiálních procesů, které jsou jiné než „běžná“ intersticiální pneumonie, kryptogenní organizující pneumonie/pneumonie s bronchiolitis obliterans, IPF, DIP a lymfocytická intersticiální pneumonie. Pacienti jsou většinou mladší než při IPF, častěji ženy a většinou nekuřačky. Bývají součástí systémových chorob pojiva, polékových změn a chronické hypersenzitivní pneumonitidy. Přítomny jsou nespecifické klinické příznaky charakteristické všem autoimunitním chorobám (horečka, slabost, váhový úbytek apod.), průběh je subakutní, podobný IPF, pouze se manifestuje v mladším věku. Na HRCT se projevuje bilaterálními, subpleurálními opacitami charakteru mléčného skla, nejčastěji v dolních lalocích. Mohou být přítomny okrsky konsolidace, kaverny jsou ale vzácné. Při biopsii plic je difuzní postižení, varianty dominantně buněčné (vzácnější) nebo fibrotizující. Oproti IPF nejsou přítomny kaverny a nález je difuzní, nikoliv fokální. Léčbou je podání glukokortikoidů + azathioprinu. Prognóza choroby je velmi dobrá (5 leté přežití > 85 %).
Podobná IPF, ale vyskytuje se v mladším věku, změny jsou difúzní a ne okrskové jako u IPF. Léčí se imunosupresí a má dobrou prognózu.
3. Akutní intersticiální pneumonie
Akutní intersticiální pneumonie (AIP, Hamman-Richův syndrom) je vzácné onemocnění probíhající většinou u osob > 40 let věku. Nespecifické prodromální příčiny se vyskytují 1 – 2 týdny před plnou manifestací choroby, následuje akutní průběh, který má podobu ARDS, s kašlem, horečkou, dušností s histologickým obrazem difuzního alveolárního poškození a hypoxémií s respiračním selháním. Na RTG S+P jsou přítomny oboustranné difuzní opacity.

Na HRCT se projevuje difuzními oboustranně symetrickými infiltráty charakteru mléčného skla, které jsou dominantně subpleurálně s možným obrazem konsolidace. Jedinou možností potvrzení diagnózy je biopsie plic s nálezem difuzního alveolárního poškození. Tedy:

Diagnóza = idiopatický ARDS + histologický průkaz difuzního alveolárního poškození
Indikována je symptomatická a resuscitační léčba, často s nutností UPV. Prognóza je špatná, i přes léčbu je 6 měsíční přežití < 40 %. Může dojít i k rekurenci choroby. U pacientů, kteří přežijí, dochází ke zlepšení plicních funkcí.
4. Kryptogenní organizující pneumonie
Kryptogenní organizující pneumonie (COP) je chorobou neznámé etiologie (vzniká idiopaticky nebo v rámci systémových chorob, po virových infekcích a po Tx plic nebo kostní dřeně), která postihuje nejčastěji jedince > 50 let. Projevuje se chřipkovými příznaky jako kašel, horečka, slabost a ztráta hmotnosti s průkazem hypoxémie a respiračního selhání. Při fyzikálním vyšetření lze nalézt inspirační chrůpky, při funkčním vyšetření restriktivní ventilační porucha. Na RTG S+P oboustranné, zřetelné, nerovnoměrné nebo difuzní alveolární opacity, mohou být rekurentní nebo migrovat. Na HRCT jsou oblasti konsolidace s opacitami mléčného skla se zesílením stěny bronchů a jejich dilatací, změny jsou nejvýraznější v bazálních partiích plic a na jejich periferii. Při biopsii plic lze prokázat granulační zánět malých dýchacích cest se zánětem přilehlých alveolů.
{kind=link}
Terapií volby jsou glukokortikoidy. Prognóza je poměrně dobrá, > 60 % pacientů zareaguje příznivě na léčbu.
Vzniká idiopaticky, u systémových chorob, po Tx plic a po virových infekcích, histologicky odpovídá granulačnímu zánětu, léčbou volby jsou glukokortikoidy.
5. Plicní alveolární proteinóza
Plicní alveolární proteinóza (PAP) je choroba postihující nejčastěji osoby ve věku 30 – 50 let, více muže. Podstatou choroby je pravděpodobně přítomnost protilátek IgG proti GM-CSF (granulocyte – macrophage colony stimulating factor) s následnou poruchou funkce alveolárních makrofágů, která vede k akumulaci PAS pozitivní lipoproteinového materiálu (surfaktantu) v alveolech.

{kind=link}
Není přítomen ani zánět, ani nedochází k poškození plicního parenchymu. Dle příčiny lze rozdělit na primární PAP s AR dědičností a sekundární PAP, který je u dospělých vzácná (intolerancí lysinurického proteinu, akutní silikózou, určitými syndromy imunodeficience, malignitami (většinou hematologickými), poruchami hematopoézy). Průběh choroby je zákeřný, projevuje se námahovou dušností, subfebriliemi, slabostí, ztrátou hmotnosti a kašlem (většinou neproduktivní, ale příležitostně může docházet k expektoraci hustého gelatinózního materiálu). Laboratorně lze nalézt polycytémii, elevaci LDH a gamaglobulinu, dále zvýšenou sérovou hladinu proteinu A a proteinu D (plicního surfaktantu), u primární formy je zvýšený titr anti-GM-CSF. Při BAL koreluje titr anti-GM-CSF v BAL se závažností choroby lépe než v séru. Na RTG S+P lze nalézt bilaterální symetrické infiltráty ve středních a dolních plicních polích, při HRCT opacity mléčného skla se zesílením interbronchiálních sept. Léčbou je laváž celých plic přes dvouluminální endotracheální trubici.
Přítomny protilátky IgG proti GM-CSF, s následnou akumulací látky podobné surfaktantu.
6. Plicní lymfangioleimyomatóza
Definice – plicní lymfangioleimyomatóza (PLAM) je vzácné multisystémové onemocnění, které se vyskytuje sporadicky nebo bývá spojováno s tuberózní sklerózou (TSC). V naprosté většině případů postihuje premenopauzální ženy. Onemocnění je charakterizováno proliferací abnormálních buněk hladkého svalstva v plicích (LAM buňky), která vede k útlaku cév a bronchů.
Choroba by měla být zvážena u mladých žen s „emfyzémem“, opakovaným pneumothoraxem a chylózním pleurálním výpotkem.
Epidemiologie – prevalence je 1:400 tisíc, většinou u premenopauzálních žen bílé rasy. Choroba progreduje během těhotenství a progrese se zpomaluje po ovarektomii.
Patogeneze – je přítomna proliferace atypických plicních hladkých svalových buněk, u kterých dochází k mutaci a získávají tak nádorový fenotyp (LAM buňky). Tvoří pak tzv. clustery v lymfatických uzlinách, kde se akumulují v jejich hilu.
Klinický obraz – projevuje se kašlem, dušností, bolestí na hrudníku pleurálního charakteru v důsledku spontánního pneumothoraxu (50 % pacientů, často s nutností pleurodézy) a/nebo chylothoraxu (také chyloperitoneum, chyloperikard a chylurie). Častěji se u těchto pacientů objevují meningeomy a angiomyolipomy ledvin (hamartomy). Život ohrožující komplikací může být hemoptýza,
Diagnostika –
Diagnóza = HRCT + biopsie (LAM buňky) a/nebo anamnéza (chylothorax/chyloperitoneum + angiomyolipomy + lymfangioleiomymom event. infiltrované uzliny/ (tuberózní skleróza/ zvýšení VEGF/pneumothorax)

Při každém podezření na PLAM musí být provedeno HRCT plic. Typická je přítomnost cyst. Všichni pacienti s PLAM nebo podezřením na ni by měli mít CT břicha (přítomnost angiomyolipomů, lymfangioleiomyomů nebo lymfadenopatie) a MR mozku při podezření na meningeom.

{kind=link}
Funkční vyšetření plic může ukázat obstrukční nebo smíšená ventilační porucha a zhoršení difuze (TLCO). Monitorace FEV1 a TLCO slouží k hodnocení odezvy na léčbu a včasnému zachycení progrese nemoci. Vhodná je i spiroergometrie a 6 minutový test chůzí k posouzení progrese nemoci. Při biopsii plic je nález LAM buněk a pozitivní imunoreaktivitou hladké svaloviny na HMB-45 (Human Melanoma Black – monoklonální protilátka reagující s antigenem vyskytujícím se v buňkách melanomu).

{kind=link}
CAVE U každého pacienta s PLAM je nutné vyloučit tuberózní sklerózu.
Terapie
- Všeobecná opatření – kontrola hmotnosti + nekuřáctví, dechová rehabilitace, bronchodilatační terapie, očkování proti chřipce + pneumokokům a léčba osteoporózy. CAVE Zcela vysadit hormonální léčbu (antikoncepci apod.), která obsahuje estrogeny, v těhotenství je potřeba zvláštní přístup (genetická konzultace, zvážení rizik apod.),
- Farmakoterapie – dosud žádná terapie neprodloužila přežití, u pacientů s rychlou progresí lze zkusit progesteron nebo sirolimus.
- Léčba komplikací (PNO, chylothoraxu, angiomyolipomů).
- Transplantace plic
Prognóza
Choroba má progresivní charakter s mediánem přežití 8 – 10 let od stanovení diagnózy.
Blíže v doporučených postupech ČPFS, dostupných na http://www.pneumologie.cz/guidelines/.
7. Deskvamativní intersticiální pneumonie
Deskvamativní intersticiální pneumonie (DIP) se vyskytuje téměř výlučně u kuřáků cigaret ve věku nejčastěji mezi 40 – 50 let. Projevuje se dušností a kašlem, poslechově jsou slyšitelné inspirační krepitace. Laboratorně zle prokázat hypoxémii, při funkčním vyšetření restriktivní ventilační porucha se snížením difuzní kapacity plic (DLCO), na zobrazovacích metodách jsou – oboustranné, difuzní, mlhavé infiltráty. Při biopsii plic lze nalézt nadměrnou akumulaci makrofágů uvnitř alveolů, často s pigmentacemi následkem kouření, jen s minimální fibrózou.

_2.jpg){kind=link}
Základním opatřením je zanechání kouření, poté je prognóza příznivá s 10 letým přežitím cca 70 %.
Choroba kuřáků cigaret, většinou středního věku. Projevuje se dušností, kašlem, inspiračními krepitacemi. Funkčně restrikce a redukce difúze. Histologicky nadměrná akumulace makrofágů v alveolech s jen mírnou fibrózou. Základním opatřením je zanechání kouření, poté je prognóza příznivá.
8. Granulomatóza z Langerhansových buněk
Granulomatóza z Langerhansových buněk (PHLB) je názvem pro škálu onemocnění charakterizovaných infiltrací různých orgánů proliferujícími Langerhansovými buňkami (LB). Jde o vzácné onemocnění s prevalencí 1:400 tisíc. Většinou se vyskytuje u mladších kuřáků cigaret (20 – 40 let), 4x častěji u mužů. Příčina je neznámá, předpokládá se, že jde reaktivní proces, vyvolaný kouřem z cigaret u predisponovaného jedince. Projevuje se dušností, kašlem, bolestivostí na hrudníku, váhovým úbytkem a horečkou. U 25 % pacientů vzniká pneumothorax. Při funkčním vyšetření plic lze nalézt restriktivní ventilační poruchu různé závažnosti a snížení difuzní kapacity plic (DLCO). RTG S+P záleží na stádiu choroby, obvyklé jsou mikronodulace, retikulace a cystické léze převážně v horních a středních plicních polích (kostofrenické úhly jsou ušetřeny). Na HRCT je kombinace uzlů, kavitovaných uzlů (tlustostěnné cysty) a tenkostěnných cyst. Hilová a mediastinální lymfadenopatie není obvyklá. Při bronchoskopii je normální obraz dýchacích cest. Při BAL převládají makrofágy. Poměr CD4/CD8 je snížený (jak je obvyklé u kuřáků). Diagnostický je> 5 % LB buněk (CD1a+) v BAL. Transbronchiální biopsie je diagnostická jen v cca 25 % (nízká senzitivita díky nerovnoměrné distribuci plicních lézí). CAVE Vyšší riziko PNO.
Radiologické nálezy zde: https://radiopaedia.org/articles/pulmonary-langerhans-cell-histiocytosis
Zásadním opatřením je zákaz kouření (pak dochází ke zlepšení klinického nálezu u 1/3 pacientů, u zbývajících 2/3 choroba postupně progreduje). V počátečních fázích onemocnění jsou indikovány glukokortikoidy (prednison 0,5 mg/kg). Při multisystémovém postižení lze přidat další imunosupresiva. PNO je léčen obvyklým způsobem. Tx plic je indikována při zhoršení respirační insuficience, často spolu s plicní hypertenzí. Recidiva v transplantované plíci je možná. Mortalita na respirační selhání je cca 10 %.
Vzácná nemoc mladších jedinců spíše mužů, kuřáků cigaret, jako atypická reakce na cigaretový kouř. Projevuje se dušností a kašlem, 25 % má PNO jako komplikaci. Postiženy jsou horní a střední plicní pole. Diagnostickým nálezem při BAL je přítomnost LB (CD1a+). Mimo omezení kouření je základní léčbou imunosuprese, v případě terminálního respiračního selhání Tx plic.
9. Léky indukované intersticiální plicní procesy
Řada léků nebo může způsobit intersticiální plicní proces, který se manifestuje dušností a suchým kašlem. Závažnost plicního procesu často závisí na dávce. Léčbou je redukce dávek nebo úplné vysazení léku:
- aspirace nosních kapek a minerálních olejů
- nitrofurantoin
- zlato, salicyláty, penicilamin
- amiodaron (často i několik let po začátku užívání)
- kyslík, talek
- bleomycin, mitomycin, cyklofosfamid, busulfan, karmustin (často i několik let po vysazení léků)
- paraguat