Hemolytické anémie
Definice – dle mechanismu vzniku jsou anémie tradičně děleny do tří skupin:
- snížená produkce erytrocytů
- jejich zvýšená destrukce erytrocytů – anémie při zvýšené destrukci je způsobena nadměrnou spotřebou erytrocytů při jejich normální (většinou ovšem kompenzatorně zvýšené) dodávce z kostní dřeně. Tento podtyp se dále dělí na:
- hemolytické anémie, při které jsou erytrocyty likvidovány uvnitř krevního oběhu.
- krvácení , kdy erytrocyty opouští krevní oběh extravaskulárně (vnitřní) nebo mimo organismus (vnější).
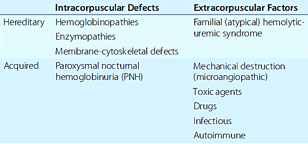
Klasifikace – hemolytické anémie lze dělit podle různých hledisek, např. vrozené / získané, akutní / chronické, intra- / extrakorpuskulární nebo intra- / extravaskulární.
Obecné klinické a laboratorní příznaky
1. Rychlost vzniku – akutní (např. autoimunitní hemolýza, favismus), chronická (mírná hereditární sférocytóza, chladové glutininy). Při chronickém trvání má organismus vůči pomalu vznikající anémii velkou adaptační kapacitu.
2. Fyzikální vyšetření – hlavním příznakem je ikterus a tmavé zbarvení moče (nekonjugovaná hyperbilirubinémie), v případě extravaskulární hemolýzy splenomegalie (v tomto případě je hlavním místem hemolýzy slezina). U závažných vrozených forem hemolytické anémie bývají deformace skeletu pro hyperaktivitu kostní dřeně (tyto změny ovšem nebývají tak těžké jako u talasémií).
3. Laboratorní nálezy
- obecné laboratorní příznaky vznikají vlastní hemolýzou a zvýšením erytropoézy v kostní dřeni – ↑ nekonjugovaný bilirubin, AST, urobilinogen ve stolici i moči (dochází ke zvýšené degradaci hemu).
- Při intravaskulární hemolýze jsou další specifické příznaky: ↑ hemoglobin v moči i séru, ↑ LDH, ↓ haptoglobin (protein, který váže volný hemoglobin). CAVE Bilirubin bývá normální nebo jen lehce zvýšený.
- Hlavním příznakem zvýšení erytropoézy je ↑ retikulocytů (jak absolutně, tak %), který je provázen ↑ MCV, polychromázie makrofágů a někdy přítomnost jaderných červených buněk při nátěru periferní krve. Aspirační biopsie kostní dřeně většinou není k diagnostice třeba, pokud se provede je typickým nálezem hyperplázie erytroidní řady.
Patofyziologie – erytrocyt během své diferenciace akumuluje ve své cytoplazmě obrovské množství hemoglobinu (zralá buňka až 340 g/l) a postupně ztrácí všechny organely i schopnost biosyntézy. Ve finálních stádiích dochází dokonce k jakési obdobě apoptózy s pyknózou až celkovou ztrátou jádra. Zralý erytrocyt následně žije průměrně 120 dnů. Červené krvinky mají výrazně redukované možnosti intermediárního metabolismu. Se ztrátou mitochondrií mizí schopnost oxidativní fosforylace, která je zprostředkovaná cytochromy a díky nepřítomnosti ribozomů není možnost proteosyntézy a tak i případné reparace poškozené buňky. Erytrocyty jsou proto velmi náchylné vůči poškození a snadno dochází k jejich destrukci. Znakem starých erytrocytů je akumulace různých membránových poškození, jejichž součástí je vazba hemichromů na intracelulární domény membránového proteinu band 3 s jejich následným shlukováním. Na něj se vážou protilátky anti-band 3 Ig (přítomné u většiny jedinců) a C3 složka komplementu. Takto opsonizované erytrocyty jsou následně vychytávány a fagocytovány retikulo-endoteliálním systémem.
Obecně lze říct, že hemolýza se manifestuje anémií, jestliže míra hemolýzy převýší produkční kapacitu kostní dřeně. Při hemolytické anémii se tak výrazně urychluje celkový obrat erytrocytů se zkrácením průměrné délky přežívání erytrocytu:
- při chronické intravaskulární hemolýze vzniká hemoglobinurie s následnou sideropenií
- naopak, při extravaskulární hemolýze (častější) dochází k přetížení železem a sekundární hemochromatóze, zejména pokud je nutnost časté aplikace transfuzí
Pokud má hemolýza dlouhodobý charakter, zvyšuje se produkce bilirubinu s predispozicí k vzniku pigmentových žlučových kamenů. Pokud je hlavním místem hemolýzy slezina, bývá často přítomna splenomegalie s příznaky hypersplenismu a související neutropenií a/nebo trombocytopenií.
Zlatým standardem je vyšetření 51Cr značenými erytrocyty, které se aplikují pacientovi, a následně se monitoruje rychlost poklesu radioaktivity. Toto vyšetření je v současné době dostupné jen v několika centrech a je zřídka indikované.
Kompenzovaná hemolýza vs hemolytická anémie – destrukce erytrocytů je silným stimulem k produkci erytropoetinu (EPO) ledvinami s následným zesílením erytropoézy. Tento mechanismus je tak silný, že je v řadě případů dostačující k adekvátní náhradě ztracených erytrocytů (kompenzovaná hemolýza) a hemolýza se neprojevuje anémií! Při objevení různých vyvolávajících faktorů (např. těhotenství, deficit folátu, renální selhání se selháním produkce EPO, infekce, deprese erytropoézy apod.) přestane erytropoéza svojí kapacitou stačit k doplnění erytrocytů v oběhu s rozvojem anémie (hemolytická anémie). Nejdramatičtější průběh mívá infekce parvovirem B19, která může vést k dramatickému poklesu erytropoézy s rychlým poklesem hemoglobinu (aplastická krize). Logicky, každý pokles erytropoézy kostní dření má u pacientů s hemolýzou dramatičtější následky, než u zdravých jedinců.
Vrozené hemolytické anémie
1. Porucha membránového cytoskeletového komplexu
Strukturální poruchy membránového cytoskeletu (nejčastěji vrozené) bývají velmi často provázeny hemolýzou. Před vlastní hemolýzou vykazují erytrocyty velmi často různé morfologické abnormality (odtud např. názvy hereditární sférocytóza nebo eliptocytóza).
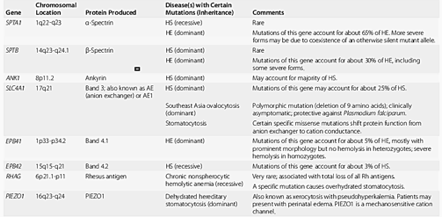
Hereditární sférocytóza
Epidemiologie – nejčastější typ vrozené hemolytické anémie s incidencí 1:5000.
Patofyziologie – chorobu poprvé popsali Minkowski a Chauffard na konci 19. století. Většinou bývá AD dědičná (nejzávažnější formy AR). Erytrocyty jsou velmi citlivé vůči hypotonickým látkám, hlavním diagnostickým kritériem je tak i přítomnost osmotické fragility. Různá závažnost klinických příznaků je způsobena heterogenitou. V mírných případech bývá hemolýza kompenzovaná a projevuje se pouze při přítomnosti zhoršujících faktorů (např. těhotenství, infekce).
Klinický obraz – různá závažnost od mírné hemolýzy po těžkou hemolytickou anémii. Hlavním příznakem bývá ikterus, splenomegalie a častá cholecystolitiáza. Často je pozitivní rodinná anamnéza hemolytické choroby, ale ne vždy (de novo mutace, recesivní forma choroby).
Diagnostika – dominují příznaky extravaskulární hemolýzy:
1. Krevní obraz
- normocytární a normochromní anémie
- zvýšení MCHC (koncentrace hemoglobinu v erytrocytech), zejména u poruch trvajících dlouhou dobu (slezina je místem hlavní destrukce, pokud ale erytrocyty průchod slezinou „přežijí“, stávají se více sférickými)
2. Nátěr periferní krve – typický nález:
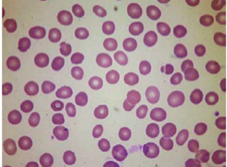
3. Ostatní – při podezření na recesivní formu choroby je třeba doplnit další testy, např. osmotickou fragilitu, lytický test s kyselinou glycerolovou, EMA (eosin-5´- maleiamid) – binding test, elektroforézu membránových proteinů na SDS gelu. Někdy dá definitivní odpověď genetické vyšetření.
Terapie – kauzální léčba neexistuje. Dříve byla základní metodou léčby splenektomie, která je nyní (pro závažné důsledky) vyhrazena jen pro těžké případy se snahou o její oddálení nejméně do věku 4 – 6 let (optimálně do puberty). Před splenektomií je jednoznačně doporučována vakcinace proti obaleným mikroorganismům (zejména pneumokokům), profylaxe penicilinem po výkonu je kontroverzní. Podobně i cholecystektomie by neměla být paušálním výkonem.
Hereditární eliptocytóza
Jak z genetického, tak z klinického pohledu velmi heterogenní choroba, která je pojmenována podle charakteristického tvaru erytrocytu, přičemž platí, že množství eliptocytů není přímo úměrné klinické závažnosti choroby. Klinický obraz i léčba jsou stejné, jako při hereditární sférocytóze. Prevalence choroby je podobná jako u hereditární sférocytózy s jednou výjimkou:
Delece devíti aminokyselin v genu SLCA4 (kóduje band 3 protein) působí chorobu zvaná ovalocytóza Jihovýchodní Asie a má v určitých populacích prevalenci více než 7 % (pravděpodobně důsledkem malarické selekce). Heterozygotní formy bývají asymptomatické, homozygotní letální.
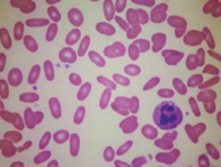
2. Poruchy transportu kationtů
Vzácné choroby, které jsou AD dědičné a jsou spojeny se zvýšením intracelulární retence sodíku a exkrece draslíku (důvod proč, se mohou projevovat pseudohyperkalémií). Následná hemolýza může mít různou závažnost. Poruchy se mohou rozdělovat do dvou kategorií:
1. Hyperhydratace erytrocytu a tudíž pokles MCHC. Na nátěru periferní krve jsou typicky přítomny lineárními erytrocyty s centrálním vyblednutím (stomatocyty). Spojena s mutací různých genů kódujících transportéry solutů, např. SLC4A1 (band 3 protein), Rhesus gen RHAG, SLC2A1 (glukózový transportér, jeho mutace je odpovědná za speciální formu zvanou kryohydrocytóza).
2. Dehydratace erytrocytů se vzestupem MCHC, erytrocyty jsou rigidní (xerocyty). Choroba vzniká mutací PIEZO1, genu pro kationický transportér.
CAVE Při stomatocytóze je splenektomie přísně kontraindikována, protože po jejím provedení mohou následovat těžké tromboembolické komplikace.
3. Porucha tvorby hemoglobinu (blíže viz kapitola 95)
Beta-talasémie
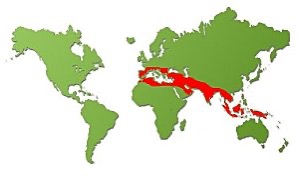
Definice – defekt syntézy beta řetězce vede k nadbytku non-beta řetězců, které nejsou rozpustné a působí oxidaci a hemolýzu erytrocytů. Erytropoéza je tak výrazně inefektivní sw vznikem anémie. Incidence je regionálně závislá (nejvíce Středozemní moře, Blízký Východ, Indie, JV Asie), v postižených regionech viz níže heterozygoti 1:100, homozygoti 1:2000). U nás jsou běžné lehké heterozygotní formy.
Etiologie a patogeneze – beta globinový řetězec je kódován na 16. chromozómu, přičemže je popsáno > 100 mutací (některé s alespoň částečně zachovalou syntézou beta řetězce – β+, jiné, u kterých je syntéza zcela potlačena – β0). Choroba je AR dědičná, s variabilní penetrancí.
Porucha tvorby beta řetězců vede k nadbytku tvorby řetězců gama a delta. Monomery hemoglobinu jsou málo rozpustné a působí poškození membrány erytrocytu s jeho hemolýzou, navíc je výrazně inefektivní erytropoéza – tvoří se méně erytrocytů, které jsou navíc nestabilní. Důsledkem je kompenzatorní hyperplázie erytropoézy se ztenčením kortikální kosti s deformacem kostí až vznikem extramedulární hematopoézy s hepatosplenomegalií. Díky hemolýze a častým transfuzím je časté přetížení železem.
Klinický obraz – velmi variabilní formy:
- minima – pouze laboratorní změny, bez anémie a klinických příznaků (běžné i u nás)
- minor – lehká anémie a splenomegalie (většinou heterozygoti β+)
- intermedia (heterozygoti β0, homozygoti β+, heterozygoti β0/ β+). Středně těžká anémie, hepatosplenomegalie, občas převody krve.
- major – homozygoti β0, těžká mikrocytární anémie, hepatosplenomegalie, deformity skeletu, již v dětství známky přetížení železem
Diagnostika – u nejzávažnějších forem je přítomna těžká mikrocytární, hypochromní anémie, zvýšené retikulocyty, bilirubin i železo a feritin v séru. Při elektroforéze hemoglobinu jsou zvýšené hladiny HbA2 a HbF. Vyetření krevního nátěru lze prokázat leptocyty (tenké erytrocyty s precipitovaným hemoglobinem centrálně – odtud název terčovité erytrocyty).
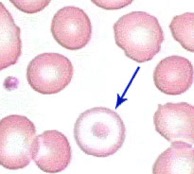
U méně závažných forem je typická mikrocytóza a hypochromie a zvýšení hladiny HbA2, méně HbF.
Terapie – u závažných forem nutné transfúze a chelatační léčba, thalassemia major je indikací k transplantaci krvetvorných buněk. Paliativně lze ke zmírnění anémie podat hydroxyureu (stimulují tvorbu HbF). Lehčí formy zpravidla nevyžadují transfúze, nutné je sledování zásob železa se včasným zahájením chelatační léčby.
Alfa-talasémie
Definice – defekt syntézy alfa řetězce vede k nadprodukci nestabilního hemoglobinu H (tetramer beta řetězců) s evznikem hemolýzy.
Etiologie – alfa řetězec je kódován dvěma páry řetězců na chromozómu 11. Mutace jednoho nebo dvou genů vede k mírné anémií, mutace tří genů vede k těžké hemolýze, mutace všech genů vede k úmrtí. U novorozenců dochází ke vzniku tetramerů řetězců gama (Hb Barts), u dospělých tetramerů řetězců beta (HbH), který je nestabilní a vede k hemolýze
Klinický obraz – heterozygoti trpí jen mírnou anémií, u homozygotů je výrazná anémie a hepatosplenomegalie.
Diagnostika – u homozygotů je těžká hypochromní anémie s bazofilním tečkováním a leptocyty. Při elektroforéze hemoglobinu je přítomný HbH a stopy Hb Barts. U heterozygotů je nález mírnější, je přítomno pouze malé množství HbH.
Terapie – u závažných forem nutné transfúze, chelatační léčba event. transplantace krvetvorných buněk.
Srpkovitá anémie
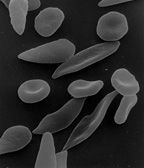
Etiologie a patogeneze – AD dědičný defekt hemoglobinu (HbS), který vzniká substitucí polární kyseliny glutamové nepolárním valinem v 6. pozici beta řetězce globinu. Takový hemoglobin je sníženě rozpustný s tendencí k polymerizaci a gelifikaci v deoxygenovaném stavu, což deformuje erytrocyt do tvaru srpku, který podléhá snadno hemolýze (extra- i intravaskulární) se zkráceným přežíváním erytrocytů.
Choroba je endemická v černošské populaci Afriky (heterozygoti tvoří 20 – 40 % obyvatelstva) a Ameriky (heterozygoti cca 8 %).

Klinický obraz
- homozygoti – HbS > 50 % s hemolýzou, která se zvyšuje při stavech se zvýšenými nároky na kyslík (infekce, prochlazení). Hemolýza je částečně extravaskulární se vznikem splenomegalie, částečně intravaskulární, kdy v oblastech se sníženou tenzí kyslíku dochází k okluzi malých cév (okluze srpkovitými erytrocyty, poškození endotelu) – vazookluzivní krize. Dochází k infarktům dlouhých kostí, kožním ulceracím, infarktům sleziny a plic a hemoglobinurii.
- heterozygoti – mírná hemolytická anémie s retikulocytózou.
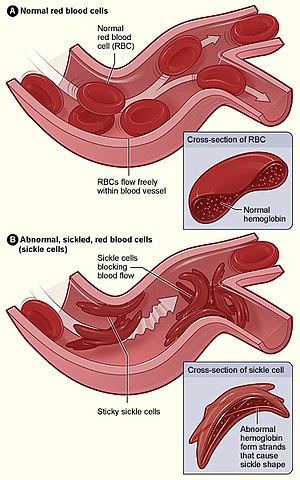
Diagnostika – diagnóza je stanovena na základě přítomnosti anémie s vyšším počtem retikulocytů, typického tvaru erytrocytů a přítomnosti intermitentních ischemických bolestí. K potvrzení slouží elektroforéza hemoglobinu.
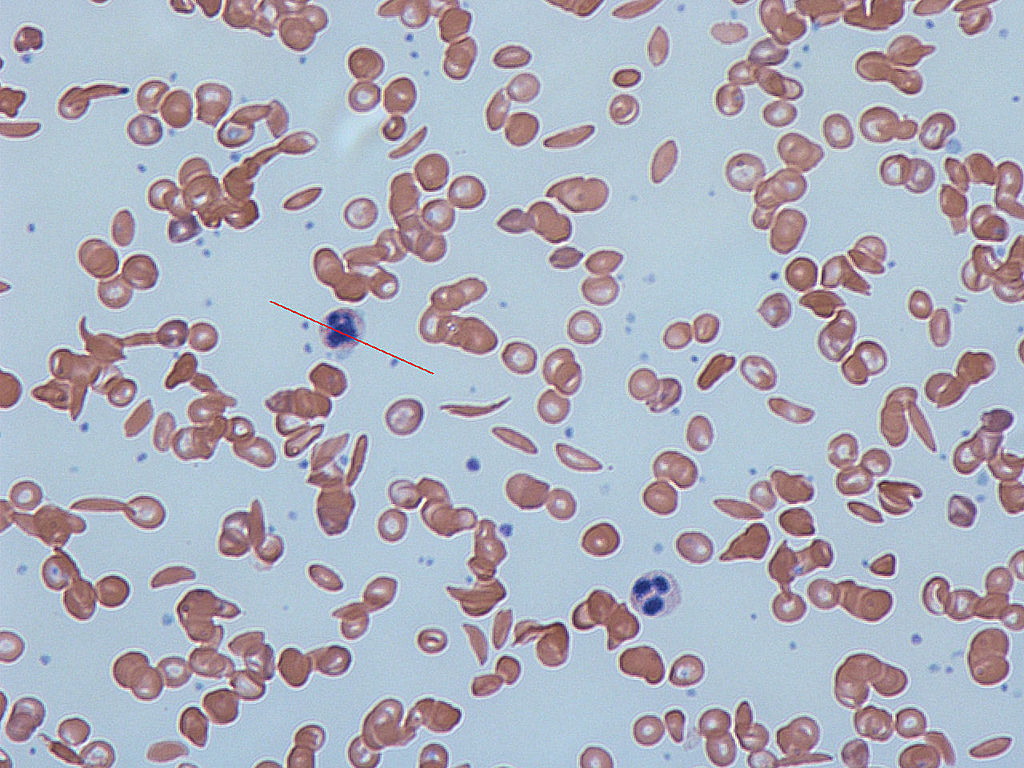
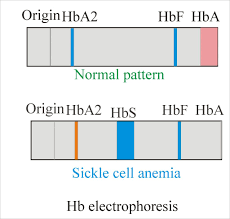
Terapie – základem je prevence vzniku stavů s absolutním nebo relativním nedostatkem kyslíku (infekce, prochlazení, dehydratace, hypoxie). Při masivní hemolýze je nutná výměnná elektrocytaferéza s transfuzní léčbou, aplikace kyslíku, hydratace s forsírováním diurézy, antkoagulační léčbou a podáváním analgetik. U homozygotů je indikována transplantace krvetvorných buněk.
Hemolgobin Lepore – genetická porucha častá ve Středozemí. Vzniká fúzní hemoglobin Lepore (α2δβ) s fenotypem podobným beta talasémii.
Hemoglobin E – HbE (α2β226Glu->Lys) je velmi častý v Jihovýchodní Asii. Je méně stabilní a klinický obraz připomíná lehčí formy beta talasémie.
4. Abnormality enzymů
Nejzávažněji se manifestují poruchy enzymů, které zajišťují energii (ATP) a/nebo zabraňují oxidačnímu poškození hemoglobinu (NADPH). Choroby lze rozdělit podle poškozené metabolické cesty a konkrétního enzymu. Jak již bylo zmíněno, v erytrocytech chybí řada organel (jádro, ribozómy i mitochondrie), jejich energetický metabolismus je tedy odkázán pouze na anaerobní glykolýzu. Defekt enzymů této metabolické cesty většinou ústí v hemolytickou chorobu.
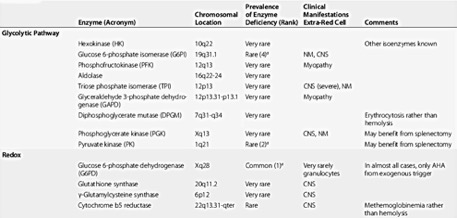
Deficit pyruvát kinázy
Epidemiologie – vrozená choroba s prevalencí 1 : 10000.
Nedávno byla u některých afrických populací nalezena polymorfní mutace genu pro pyruvát kinázu (E277K) s prevalencí 1 – 7 %, pravděpodobně opět díky malarické selekci.
Patofyziologie – nedostatek enzymu vede k nedostatečné produkci ATP, následkem je lýza erytrocytů nebo jejich předčasná destrukce ve slezině.
Klinický obraz
- u homozygotů se projevuje perzistencí novorozeneckého ikteru s velmi vysokou retikulocytózou.
- u heterozygotů může být diagnostika opožděna, protože anémie se často projevuje až při zhoršujícím faktoru (infekce, těhotenství). Bývá různě vyjádřena, někdy je mírná, jindy vyžaduje krevní převody. Bývá velmi dobře tolerována, protože metabolický blok se nachází v posledním kroku glykolytické cesty, se zvýšením hladiny 2.3-bisfosfoglycerátu, který posouvá disociační křivku hemoglobinu pro kyslík doprava s následným zvýšením uvolňování kyslíku ve tkáních.
Terapie – léčba je symptomatická. Vzhledem k vysokému obratu erytrocytů je třeba dostatečná substituce kyseliny listové. Při potřebě jsou indikovány krevní transfúze, pro riziko přetížení železem často s nutností chelatační léčby. Při těžkém průběhu může být indikována splenektomie. U matek, které mají již jedno postiženě dítě, je indikován prenatální screening.
Byl popsán jediný případ vyléčení choroby transplantací kostní dřeně od HLA identického příbuzného. U myšího modelu byl úspěšně použit transfer genů pomoci lentivirového vektoru. Defekt ostatních enzymů glykolytické cesty je velmi vzácný a v důsledku také působí hemolýzu. Anémie bývá různě závažně vyjádřena, často je přítomna splenomegalie. U některých defektů je současně postižen neuromuskulární a//nebo centrální nervový systém (např. deficit trióza fosfát izomerázy). Diagnóza hemolýzy není obtížná (normo- nebo makrocytární anémie + retikulocytóza + hyperbilirubinémie). Coombsův test bývá negativní. Diagnóza je potvrzena genetickým vyšetřením. V jednom případě deficitu fosfoglycerát kinázy byla allogenní transplantace kostní dřeně úspěšná v kontrole hemolýzy, ale již nezvrátila neurologické poškození.
Deficit glukóza-6-fosfát dehydrogenázy
Epidemiologie – široce rozšířeno v tropickém a subtropickém pásu (Afrika, Jižní Evropa, Středí Východ, Jihovýchodní Asie a Oceánie), v některých oblastech je jeho prevalence 20 % i více. Tato regionální diskrepance je pravděpodobně způsobena faktem, že erytrocyty s deficitem G6PD jsou rezistentnější vůči malárii způsobené Plasmodium falciparum. Odhaduje se, že celosvětově je postiženo 400 miliónů jedinců.
Regionálně lze rozlišit několik variant této choroby: Mediterranean – Středozemní moře, Střední Východ, Indie, Varianta A – Afrika, Jihovýchodní Evropa, Vainchan, Mahidol – Jihovýchodní Asie, Canton – Čína, Union – široce rozšířené
Etiologie – gen pro G6PD je vázaný na X chromozóm. Muži mají tak pouze jeden G6PD gen (jsou hemizygoti) a proto musí být buď pouze zdraví, nebo postižení. Naopak ženy mohou být i heterozygoti a choroba se u nich může projevit střední závažností. Heterozygotní ženy mohou mít genetický mozaicismus s velmi variabilní aktivitou sledovaného enzymu. Zatím bylo popsáno cca 180 různých mutací tohoto genu.
Patofyziologie – glukóza-6-fosfát dehydrogenázy (G6PD) je enzym (dimer nebo tetramer složený ze dvou nebo čtyř podjednotek, každá o velikosti 514 aminokyselin) se zásadní funkcí oxidačně-redukčních dějích všech aerobních buněk. U erytrocytů je jeho úloha ještě důležitější, protože G6PD je u nich jediných zdrojem NADPH, který jak přímo, tak prostřednictvím glutathionu, chrání buňku před oxidačním stresem. Důsledkem mutace G6PD je ve většině případů zrychlení stárnutí erytrocytů, v některých případech poškození katalytické funkce tohoto enzymu. V ostatních tkáních je manifestace defektů méně výrazná.
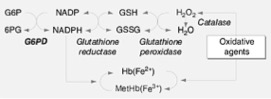
Klinický obraz – většina osob je asymptomatických. Choroba se projevuje odlišně u novorozenců a dospělých.
U novorozenců vzniká ikterus po 2 – 3 dnech od narození, zatímco anémie většinou nebývá závažná. V některých případech s vysokým oxidačním stresem (předčasně narozené děti, infekce a/nebo enviromentální faktory, např. kuličky s naftalenem a kafrem, které se někdy dávají do oblečení a postýlek) a/nebo přítomnosti monoalelické nebo bialelické mutace genu pro uridyltransferázu (UGT1A, stejný, který je zodpovědný za Gilbertův syndrom) může novorozenecký ikterus probíhat velmi závažně s rizikem neurologického postižení.
Hemolytická epizoda u dospělých typicky začíná nevolností, slabostí a bolestmi břicha a bederní krajiny. Po intervalu několika hodin až 2 – 3 dnů se rozvíjí ikterus s tmavou močí. Nástup může být extrémně rychlý, zejména u favismu dětí. Anémie bývá středně závažná až těžká, obvykle normocytární a normochromní. Při ústupu hemolýzy se kompenzuje i anémie. Nejzávažnější komplikací je vznik akutního renálního selhání, které má vynikající prognózu u pacientů s dříve zdravými ledvinami.
Oxidační stres může být vyvolán třemi způsoby:
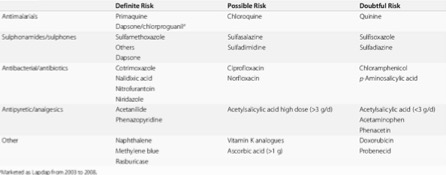
- Požití favových bobů
- Infekce
- Některé léky – problémem může být, že se oblast výskytu deficitu G6PD překrývá s malarickými regiony. Nyní dochází k návratu k aplikaci primachinu, který má jako jediné antimalarikum schopnost eliminovat gametocyty Plasmodium falciparum (tedy zabránit dalšímu přenosu) a hypnozoity Plasmodium vivax (tedy zabránit endogennímu relapsu). Plošné podání primachinu je ovšem velmi rizikové (vyvolává oxidační stres a při deficitu G6PD hemolýzu). Podobně, podání rasburikázy osobám s deficitem G6PD bylo spojeno s několika smrtelnými případy (u novorozenců s renálním selháním a dospělých s tumor lysis syndromem).
Velmi malá část pacientů s deficitem G6PD trpí chronickou nesférocytární hemolytickou anémií (CNSHA) různé závažnosti. Většinou jde o muže, často s anamnézou novorozeneckého ikteru, u kterých se choroba projevuje anémií, nevysvětlitelným ikterem a přítomností žlučových kamenů, Anémie bývá normo- nebo makrocytární s vysokou hladinou retikulocytů. Přestože je hemolýza chronická, může být dále zhoršena oxidačním stresem, proto i u CNSHA platí stejná režimová opatření jako u ostatních pacientů deficitu G6PD. V některých případech CNSHA je deficit G6PD tak výrazný, že se projevuje i u granulocytů nedostatečnou schopností oxidačního vzplanutí, což sebou nese zvýšené riziko některých bakteriálních infekcí.
Diagnostika
1. Laboratorní testy (laboratorní příznaky intra- i extravaskulární hemolýzy):
- normocytární a normochromní anémie středně závažná až závažná
- hemolýza je částečně intravaskulární, takže je přítomen i volný hemoglobin v plazmě, hemoglobinurie, ↓haptoglobin, ↑ LDH
- ↑ nekonjugovaný bilirubin dokazuje, že hemolýza je i částečně extravaskulární
2. Nátěr periferní krve – obraz anizocytózy a polychromázie s přítomností sférocytů. Nejtypičtějším znakem je přítomnost bizárních poikilocytů, které mají nepravidelně rozložený hemoglobin („hemighosts“) a vypadají, jako by byla jejich část ukousnuta pryč („bite cells“).
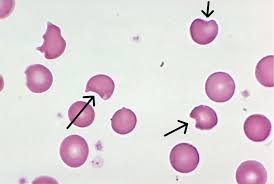
3. Genetické vyšetření
Terapie – základem léčby jsou režimová opatření. Je třeba se vyvarovat konzumaci svinského bobu (Vicia fava) i rizikových léků (před nasazením daného léku v endemické oblasti je vhodné screeningové vyšetření na přítomnost deficitu G6PD). Při rozvoji hemolýzy nebývá potřeba žádné léčby, při těžké anémii (zejména u dětí) jsou nutné krevní transfúze. Při přítomnosti akutního renálního selhání je indikována hemodialýza.
U CNSHA s chronickou hemolýzou je vhodná substituce kyseliny listové a hematologické sledování. Vzácně je hemolýza tak závažná, že jsou potřeba pravidelné krevní transfúze, často s nutností chelatační léčby k zabránění přetížení železem. Na rozdíl od hereditární sférocytózy, nejsou důkazy o selektivní destrukci erytrocytů ve slezině, přesto v některých těžkých případech byla splenektomie úspěšná.
Infantilní poikilocytóza
Mírná hemolytická anémie, která samostatně odeznívá a je způsobena nutričním nedostatkem selenu, který je zásadním kofaktorem pro činnost glutathion peroxidázy.
Deficit pyrimidin 5´- nukleotidázy
Pyrimidin 5´- nukleotidázy (P5N) je zásadní při degradaci a katabolismu nukleotidů při pyknóze jádra zrajícího erytrocytu. Podstata vzniku hemolytické anémie není příliš jasná, ale je spojena s morfologickou abnormalitou, tzv. bazofilním tečkováním. Deficit enzymu je vzácný, vzniklá anémie je doživotní, různé závažnosti a někdy může pomoci splenektomie.
5. Familiární (atypický) hemolyticko-uremický syndrom
Definice – jako atypický HUS se označuje skupina vzácných chorob, postihujících zejména děti, projevujících se mikroangiopatickou hemolytickou anémií s přítomností fragmentovaných erytrocytů + trombocytopenií (většinou mírnou) + akutním renálním selháním. Naproti tomu, typický HUS je způsoben infekcí kmenem Escherichia coli, který produkuje Shiga toxin.
Etiologie – příčina je genetická, byly prokázany mutace genů kódujících proteiny regulující komplement: faktor H (gen CFH), faktor I (gen CFI), faktor B (gen CFB), C3 složku komplementu, trombomodulin a CD46 (gen MCP).
Patofyziologie a klinický obraz – vzhledem k fyziologickému nadbytku regulačních proteinů komplementu se jejich nedostatek projevuje pouze při nadměrné aktivaci komplementu (např. interkurentní infekce) a dochází k poškození endoteliálních buněk (zejména v ledvinách) a erytrocytů se vznikem hemolýzy. Následná ataka atypického HUS má 15 % mortalitu a 50 % nemocných progreduje do end stage renálního selhání. Stav se často upravuje ad integrum, ovšem s velkým nebezpečím recidivy při objevení se dalšího triggeru aktivace komplementu. Choroba má vážnou prognózu.
Terapie – základem léčby jsou plazmaferézy, které doplňují chybějící regulační protein. Ke zlepšení stavu vede eculizumab, anti-C5 protilátka inhibující komplement. Otázkou zatím zůstává, jak dlouho ecilizumab podávat a jak výrazně ovlivňuje prognózu transplantace jater a ledvin u vybraných pacientů.
Získané hemolytické anémie
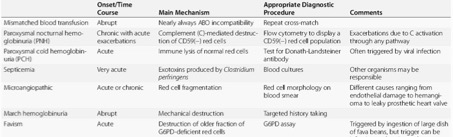
1. Mechanická destrukce erytrocytů
- Pochodová hemoglobinurie – vyskytuje se u maratonských běžců, vzácněji u bosých rituálních tanečníků nebo hráčů na bubny bongo. Není jasné, proč se u některých jedinců hemolýza vyskytne a u jiných nikoliv.
- Mikroangiopatická hemolytická anémie – chronická hemolýza vznikající u pacientů s náhradou srdeční chlopně, zejména v přítomnosti paraprotetické regurgitace. Pokud je hemolýza mírná a suplementuje se folát a železo, choroba se neprojeví. Při závažnější anémii je indikován korektivní kardiochirurgický výkon.
2. Infekce
- Malárie – v endemických oblastech je zdaleka nejčastější infekční příčinou hemolytické anémie.
- Hemolyticko-uremický syndrom – v nemalarických regionech je nejčastější příčinou infekce kmene Escherichia coli (kmen O157L:H7 produkující Shiga toxin), který je hlavním původcem hemolyticko-uremického syndomu. Choroba se vyskytuje častěji u dětí než u dospělých.
- Klostridiová sepse – klostridiová sepse (Clostridium perfringens, produkující toxin s lecitinázovou aktivitou) při infikovaných otevřených ránách a septických abortech může být provázena masivní hemolýzou.
Hemolýza může mnohem vzácněji (a téměř vždy u dětí) provázet sepse a endokarditidy všech původců. Všechny infekce jsou významnou příčinou oxidačního stresu, takže mohou být triggerem hemolýzy i u pacientů s deficiencí G6PD.
3. Imunitní hemolytické anémie
Dělení podle řady hledisek:
- podle typu protilátek (autoprotilátky proti antigenům na povrchu erytrocytu/protilátky proti antigenům mimo erytrocyt (např. léky), které se chovají jako hapteny)
- ideální teploty reakce (tepelné/chladové)
- podle původu (idiopatické / sekundární např. při SLE nebo CLL)

Autoimunitní hemolytické anémie
Patofyziologie – pokud je erytrocyt „označen“ autoprotilátkou, je následně různým mechanismem zlikvidován s rozvojem autoimunitní hemolytické anémie (AIHA).
1. Extravaskulární hemolýza (makrofágy) – Fc konec autoprotilátky vázané na erytrocyt je nejčastěji rozeznán Fc receptorem makrofágů s jeho následnou fagocytózou. Hlavním místem fagocytózy jsou proto tkáně, kde se nachází velké množství makrofágů (zejména slezina, ale i játra a kostní dřeň).
2. Intravaskulární hemolýza (komplement) – v některých případech má autoprotilátka (nejčastěji IgM) vázaná na svůj antigen schopnost aktivovat komplement s následnou hemolýzou přímo v krevní cévě.

Klinický obraz
Charakteristické trias = anémie + ikterus + splenomegalie.
AIHA má bez adekvátní léčby 10 % mortalitu, často s náhlým vznikem a dramatickým průběhem. Anémie bývá závažná s poklesem hemoglobinu na 40 g/l během několika dnů, rozvojem ikteru a někdy splenomegalie. Pokud je hemolýza alespoň částečně intravaskulární, je přítomna hemoglobinurie s tmavou močí. V některých případech může být AIHA spojena i s trombocytopenií (Evansův syndrom).
Diagnostika – základem je přímý antiglobulinový test (vyvinul jej R. R. A. Coombs v roce 1945. Coombsův test přímo prokazuje přítomnost autoprotilátek vázaných na erytrocytu. V některých případech jsou definovány konkrétní protilátky, v jiných případech je test nespecifický s reaktivitou všech typů erytrocytů. Senzitivita Coombova testu je poměrně nízká.
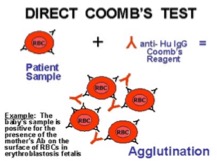
AIHA může vznikat jako komplikace systémového lupus erythematosus nebo chronické lymfocytární leukémie a proto je nutné po těchto chorobách vždy cíleně pátrat a vyloučit je.
Terapie
1. Transfúzní terapie – těžká AIHA vyžaduje akutní léčbu. Hlavní formou léčby bývá podání krevních transfúzí. To v některých případech může znamenat problém, protože pokud jsou autoprotilátky nespecifické, stává se celá krevní transfúze inkompatibilní. Pokud není anémie život ohrožující, měly by se transfúze odložit.
2. Farmakoterapie – u nejméně 50 % dochází k rychlé remisi po podání prednisonu (1 mg/kg denně). Při relapsu + anti-CD20 rituximab v nízké dávce (100 mg týdně po dobu 1 měsíce), což snižuje riziko relapsu, který je u AIHA častý.
3. Chirurgická terapie – u pacientů refrakterních k předchozím postupům je vhodné vyzkoušet splenektomii, která sice neléčí chorobu, ale odstraňuje hlavní místo, ve kterém dochází k hemolýze. Tento postup zlepšuje anémii a snižuje potřebu ostatní terapie (např. redukce dávky prednisonu). Po objevu rituximabu, intravenózních imunoglobulinů a ostatních imunosupresiv je splenektomie indikována velmi vzácně.
4. Transplantace kostní dřeně – autologní nebo alogenní transplantace kostní dřeně je velmi vzácně indikována u velmi těžkých a refrakterních případů.
Paroxyzmální chladová hemoglobinurie
Epidemiologie – velmi vzácná forma AIHA vyskytující se zejména v dětském věku.
Patofyziologie (komplement) – spouštěcím faktorem bývá virová infekce (většinou ustupuje spontánně), která indukuje vznik speciálních, tzv. Donath-Landsteinerových protilátek, které mají anti-P specifitu (nejčastěji IgG) a váží se na erytrocyty pouze za nízké teploty (optimum 4°C). Jakmile teplota vzroste na 37°C dochází za přítomnosti komplementu k intravaskulární hemolýze s hemoglobinurií.
virová infekce → D-L protilátky při 4°C na erytrocyty → aktivace komplementu s hemolýzou při 37°C
Diagnostika – diferenciálně diagnosticky se musí vyloučit ostatní příčiny hemoglobinurie, specifickým nálezem je průkaz Donath-Landsteinerových protilátek.
Terapie – hlavní je symptomatická léčba, včetně podávání krevních transfúzí.
Nemoc chladových aglutininů
Definice – CAD je typem chronické AIHA. Postihuje zejména starší jedince.
Patofyziologie – autoprotilátky produkovány specifickým klonem B-lymfocytů (anti-antigen I, většinou typ IgM → CAD se považuje za časnou formu Waldenströmovy makroglobulinémie, většinou ale chybí ostatní projevy této choroby, často mívají tak vysoký titr, že při elektroforéze séra mohou imitovat paraprotein). Reagují nejlépe při teplotě cca 4°C, při vzestupu teploty se na ně váže C3 složka komplementu s indukcí hemolýzy.
Terapie
1. Režimová opatření – u lehkých forem dodržování režimových opatření (vyvarování chladu), těžké formy jsou léčitelné velmi obtížně.
2. Transfúzní terapie – krevní transfúze jsou neefektivní (přijaté erytrocyty jsou I pozitivní a jsou rychle destruovány).
3. Farmakoterapie – imunosupresivní léčba (glukokortikoidy, azathioprin nebo cyklofosfamid) a splenektomie nejsou dostatečně efektivní a nežádoucí účinky léčby mohou převýšit její benefit. Rituximab má efekt u více než 60 % pacientů, přestože není tak účinný jako u AIHA. Remisi lze prodloužit kombinací s fludarabinem.
4. Plazmaferézy – provádění plazmaferéz má logické zdůvodnění, ale k tomu, aby měly efekt, musí být prováděny pravidelně.
4. Toxické a polékové hemolytické anémie
1. Oxidačně působící látky – řada látek, které mají oxidační potenciál, mohou způsobit hemolýzu i u osob bez deficitu G6PD (např. 100 % kyslík, nitráty, chloráty, metylenová modř, dapson, cisplatina, některé aromatické uhlovodíky).
2. Ostatní látky – jiné látky mohou působit neoxidativní hemolýzu, většinou neznámým mechanismem (např. arsen, stibin, olovo, měď). Hemolytická anémie způsobená intoxikací olovem je charakteristická bazofilním tečkováním. Podobnost s deficitem pyrimidin 5´- nukleotidázy naznačuje, že olovo tento enzym pravděpodobně inhibuje.
3. Hadí a pavoučí jedy – k těžké hemolýze dochází působením jedů některých hadů (např. kobra a zmije) a pavouků.
Patofyziologie – k hemolýze dochází dvojím mechanismem:
- Látka působí jako hapten s následnou indukcí protilátek (vzácně penicilín).
- Látka působí jako spouštěč produkce autoprotilátek proti antigenům erytrocytů (metyldopa stimuluje tvorbu Rh protilátek anti-e u malého množství pacientů. U pacientů, kteří mají antigen e ve svoji výbavě, působí hemolýzu).
5. Paroxyzmální noční hemoglobinurie
Definice – PNH je chronicky probíhající získaná hemolytická anémie, pro kterou je charakteristické trias (ne vždy se všechny složky manifestují):
Hemolýza + pancytopenie + sklon k žilním trombózám
Epidemiologie – prevalence 1: 200 000, regionálně ani mezi pohlavími se neliší (snad mírně častěji v Jihovýchodní Asii a na Dálném Východě).
Patofyziologie – nedostatek inhibitorů komplementu v membráně erytrocytu (nejdůležitější je CD59, který brání interakci s C9 (membrane attack complex) se zvýšenou citlivostí erytrocytů vůči aktivovanému komplementu (nezávisle jestli klasickou nebo alternativní cestou). Patofyziologická podstata tendence k žilním trombózám není ještě jasná, vysvětlením může být deficit CD59 na trombocytech, vedoucí k jejich nadměrné aktivaci.
Vlastní podstatou PNH je získaná X-vázaná mutace genu PIG-A působící zkrácení glykofosfatidylinositolové kotvy, která tyto membránové proteiny váže přes peptidové řetězce k cytoplazmatické membráně. Vzhledem k tomu, že nejde o vrozenou mutaci, vlastní strukturální defekt je u každého pacienta odlišný. Ve svém důsledku se v kostní dření i periferní krvi nachází jak normální, tak mutantní erytrocyty.
Pacienti s PNH mají často anamnézu aplastické anémie (AA) nebo selhání kostní dřeně (BMF) a naopak. U některých pacientů může být méně vyjádřena hemolýza a více aplázie. Obě choroby mají i podobnou patofyziologii, protože AA je autoimunitní chorobou, při které T lymfocyty likvidují krvetvorné kmenové buňky. PNH se liší tím, že cílem T-lymfocytů bývají pouze jejich PIG-A mutantní varianty.
T lymfocyty likvidují všechny erytrocyty (AA) / PIG-A mutantní erytrocyty (PNH).
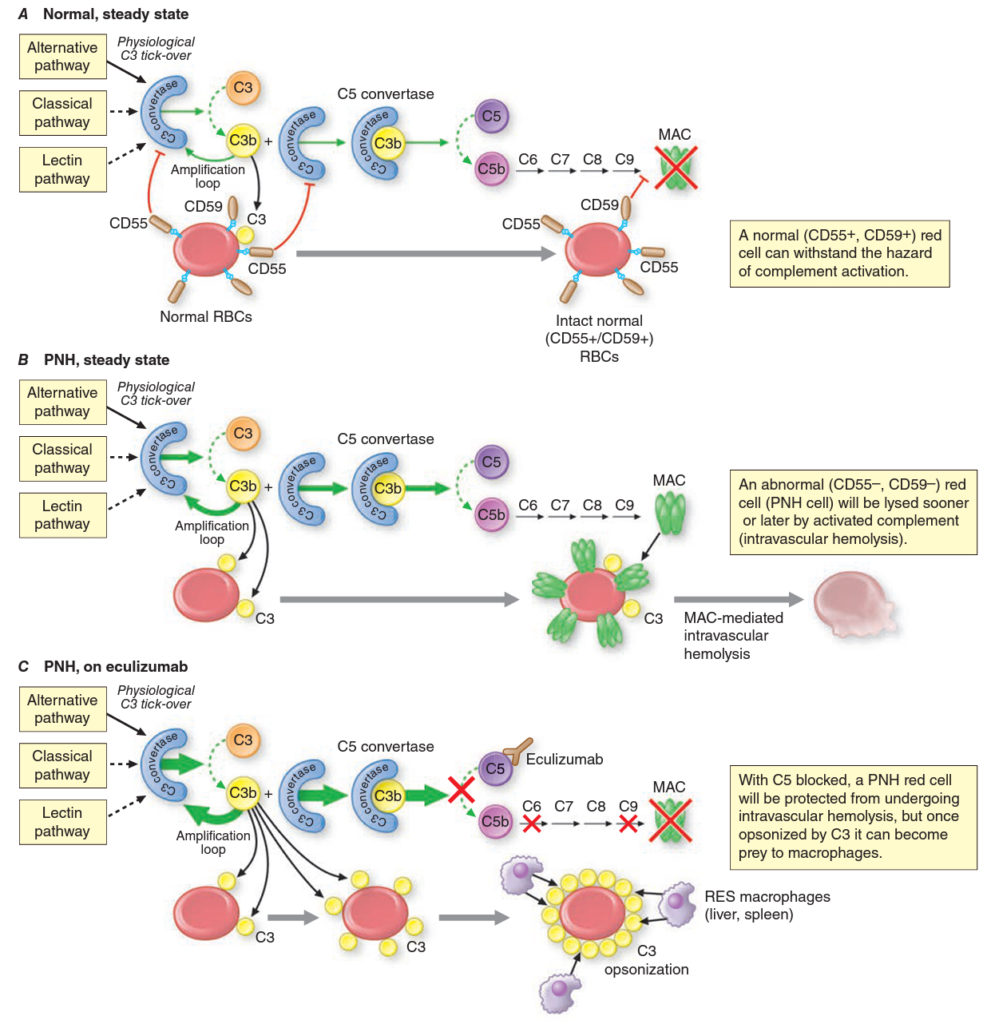
A. Normální erytrocyty jsou proti aktivovanému komplementu chráněni CD59 a CD55.
B. Stav při PNH.
C, Stav po léčbě eculizumabem (inhibitor C5a)
Klinický obraz – typická trias: recidivující hemolýza + pancytopenie + sklon k žilním trombózám (ne vždy jsou vyjádřeny všechny složky). Část pacientů vyhledá lékaře pro objevení krve místo moče, většinou jsou první příznaky méně dramatické a choroba je diagnostikována při došetřování anémie (někdy + trombocytopenií a neutropenie při selhání kostní dřeně). Někteří pacienti si stěžují na bolestivost břicha, což může souviset s trombózou viscerálních žil. U 1 – 2 % pacientů může PNH progredovat do akutní myeloidní leukémie.
CAVE Při vzniku Budd-Chiarriho syndromu v terénu normálních jater je nutné vždy vyloučit PNH.

Diagnostika
1. Krevní obraz – základním nálezem je anémie (lehká až velmi závažná), většinou normocytární a normochromní s normální morfologií. Pokud je makrocytární, bývá spojena s výraznou retikulocytózou (i více než 20 %). Při chronické ztrátě železa pro hemoglobinurii může být mikrocytární.
2. Biochemické vyšetření – nekonjugovaný bilirubin mírně nebo středně zvýšený, LDH typicky výrazně zvýšený a nedetektovatelný haptoglobin.
3. Vyšetření moče – typickým nálezem je hemoglobinurie, která se velmi rychle mění v čase, proto je vhodné odebrat sérii vzorků a až tyto analyzovat.
4. Vyšetření kostní dřeně – kostní dřeň je celulární s výraznou erytroidní hyperplázií, často s mírnou nebo střední dyserytropoézou (nezaměňovat s myelodysplastickým syndromem!). Někdy lze ale naopak nalézt její hypoplázii až aplázii.
5. Specifické testy – specifické testy prokazují zvýšenou citlivost erytrocytů vůči aktivovanému komplementu (sacharózový hemolytický test – je nespolehlivý, Hamův test – používá se acidifikované sérum, specifický, ale jen velmi málo dostupný):
Průtoková cytometrie – zlatý standard diagnostiky PNH, detekuje nepřítomnost CD59 a CD55 na povrchu erytrocytů (při PNH > 5 % všech erytrocytů nebo > 20 % všech granulocytů)
Terapie – intenzita terapie záleží na závažnosti choroby. Někdy stačí strategie „watch and wait“, jindy je třeba intenzivní léčba:
1. Podpůrná léčba – důležitá je suplementace kyseliny listové (nejméně 3 mg/den), hladinu železa je třeba monitorovat a při deficitu substituovat.
2. Farmakoterapie – glukokortikoidy nejsou doporučovány, protože nebyl prokázán žádný efekt. Základem léčby je eculizumab, lidská monoklonální protilátka, která váže na C5 složku komplementu a brání její aktivaci, která poté indukuje formaci membránového lytického komplexu. Snižuje hemolýzu a zlepšuje kvalitu života pacientů. U poloviny léčených pacientů dochází ke zvýšení hladiny hemoglobinu, u zbytku přetrvává těžká anémie s nutností krevních transfuzí. Jednou z příčin je, že při blokádě distálních partií komplementové kaskády zůstávají erytrocyty opsonizovány C3 složkou komplementu a poté hemolyzovány extravaskulárně. Rozsah této extravaskulární hemolýzy závisí na genetickém polymorfismu komplementového receptoru CR1. Vzhledem k jeho biologickému poločasu, musí být eculizumab podáván každých 14 dnů.
U pacientů s kombinací PNH-AA může být indikována kombinovaná imunosupresivní léčba s antithymocytárním globulinem a cyklosporinem, zejména v případě omezeného účinku ostatní terapie.
Antikoagulační léčba je indikována u všech pacientů s žilní trombózou, při závažným komplikacích je možné použít i trombolýzu.
3. Allogenní transplantace kostní dřeně – jediná kurativní možnost léčby u pacientů s PNH je allogenní transplantace kostní dřeně od HLA identického příbuzného. Je většinou prováděna mladým pacientům s těžkou formou PNH, od objevu eculizumabu indikována velmi vzácně.
Prognóza – medián přežití neléčených pacientů je 8 – 10 let, nejčastější příčinou smrti jsou následky žilní trombózy, následované infekcí při těžké neutropenii nebo krvácením při těžké trombocytopenii.
97. Anémie při akutních krevních ztrátách
Patofyziologie – krevní ztráty způsobují anémii dvojím způsobem, akutně ztrátou erytrocytů nebo chronickým, protrahovaným krvácením spojeným s deplecí zásob železa. Pacient může krvácet zevně (např. trauma) nebo vnitřně (např. krvácení z jícnových varixů, ruptura sleziny, extrauterinní krvácení nebo subarachnoidální krvácení). Jakékoliv akutní krvácení má několik fází:
- Akutní hypovolémie postihující hlavně orgány, které jsou bohatě zásobené krví (např. mozek nebo ledviny). Akutní krvácení se tak projevuje zhoršením vědomí a funkce ledvin.
- Stresové receptory a baroreceptory poté indukují uvolnění vazopresinu a ostatních peptidů s následným přechodem extravaskulární tekutiny intravaskulárně a hemodilucí. Díky tomu anémie vzniká až v této fázi. Pokud např. úroveň hemoglobinu po 3 dnech poklesne na hladinu 70 g/l, znamená to, že došlo ke ztrátě poloviny celkového objemu krve.
- Po ukončení krvácení se anémie postupně zmírňuje, díky dodávce nových erytrocytů z kostní dřeně.
Diagnostika – diagnostika akutní posthemoragické anémie nebývá někdy přímá, protože některé epizody se neprojeví jasnými symptomy. Při jakémkoliv náhlém poklesu hemoglobinu je proto nutné nejdříve vyloučit krvácení (pečlivá anamnéza, ultrazvukové nebo endoskopické vyšetření…).
Terapie – léčba posthemoragické anémie závisí na rychlosti jejího vzniku a na řadě dalších faktorů. Lidské tělo není na rychle vzniklou anémii dobře adaptované. Hlavním cílem je zastavení krvácení a při potřebě podání krevních transfúzí (při elektivních výkonech jsou s výhodou autotranfúze).
Nyní probíhají pokusy s roztoky, které by substituovaly lidskou krev, zejména na bázi fluorokarbonu a arteficiálně modifikované hemoglobiny (HBOCs, hemoglobin-based oxygen carriers).