U cca 1 ze 4000 novorozenců je nutné vyšetření pro atypický vývoj genitálu. U pohlaví je nutné definovat rozdíly v terminologii:
- chromozomální pohlaví – je dané karyotypem 46XY u mužů, 46XX u žen. Plody 45Y nejsou viabilní.
- gonadální pohlaví – je dáno přítomností histologické tkáně (ovarium/varlata specifické pro dané pohlaví).
I. Poruchy chromozomálního pohlaví
Klinefelterův syndrom (47, XXY)
Epidemiologie – objevuje se u mužů v četnosti cca 1:1000, ale v cca 75 % není diagnostikován (pokud ano, tak nejčastěji po pubertě).
Patofyziologie – klasická forma Klinefelterova syndromu (KS) se objevuje při meiotické nondisjunkci pohlavních chromozómů, formy 48, XXYY nebo 48, XXXY jsou vzácné.
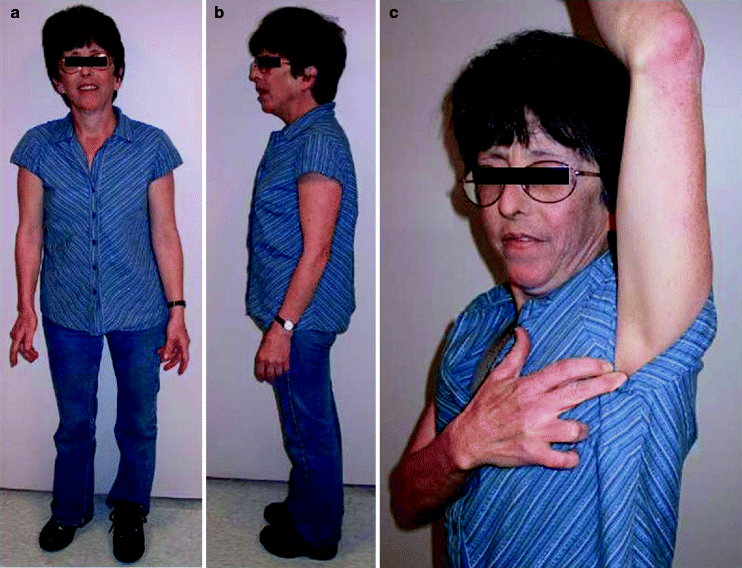
Klinický obraz – typická jsou malá varlata (nejčastěji < 2,5 cm), neplodnost (hyalinizace semenotvorných kanálků, azoospermie), gynekomastie (zvýšená hladina estrogenů), vysoká postava s dlouhými končetinami a hypogonadismus u fenotypicky mužů. Laboratorně lze prokázat zvýšenou hladinu FSH a LH a nízkou hladinu testosteronu.
Terapie – substituce androgenů zlepšuje virilizaci, libido, kostní denzitu a snižuje únavu, na druhou stranu může zhoršit gynekomastii, kterou lze ovšem řešit chirurgicky. Oplodnění partnerky lze dosáhnout IVF. U pacientů s KS je zvýšené riziko karcinomu prsu, kardiovaskulárních chorob, metabolického syndromu a autoimunitních poruch.

Turnerův syndrom (45, X)
Epidemiologie – objevuje se u žen v četnosti cca 1:2500.
Patofyziologie – 50 % žen s Turnerův syndrom (TS) má karyotyp 45,X, cca 20 % pak mozaicismus 45,X/46,XX.
Klinický obraz – u TS lze při prenatální diagnostice prokázat zvýšené nuchální projasnění, ženy mají místo ovárií vazivové proužky s následnou primární amenoreou, neplodností a nízkou výškou postavy (obvykle < 150 cm), častý je i široký krk s kožními řasami. U 30 % pacientů jsou přítomny i levostranné srdeční vady (30 – 50 % bikuspidní aortální chlopeň, 30 % koarktace aorty, 5 % dilatace kořene aorty) a u 30 % dále vrozené malformace ledvin a vývodných močových cest. V dětství jsou časté otitis media a postupem věku častější senzoneurální hluchota a primární hypotyreoidismus.
Terapie – základem je terapie jednotlivých složek syndromu. Ve věku cca 11 let je vhodné zahájení terapie nízkými dávkami estrogenů k indukci puberty a zajištění řádného pohlavního vývoje, později se přidává substituce progesteronu. U některých žen se podařilo IVF po přijetí darovaných vajíček s následným úspěšným dokončením těhotenství.

Ovotestikulární porucha sexuálního vývoje – dříve pravý hermafroditismus. Většina jedinců má karyotyp 46, XX a má přítomno nejednoznačné pohlaví.
II. Poruchy gonadálního pohlaví
Poruchy vývoje varlete (DSD) je vhodné rozdělovat podle karyotypu, protože podle fenotypu je toto dělení nejednoznačné.
46, XY DSD
I. Porucha vývoje varlat
Úplná gonadální dysgeneze (Swyerův syndrom)
Gonády jsou pouze ve formě vazivového pruhu, díky nepřítomnosti AMH/MIS jsou přítomny struktury Müllerova vývodu a úplná absence androgenizace. I přes karyotyp 46, XY je přítomen ženský fenotyp. Periferní pohlavní hormony, inhibin B i AMH/MIS jsou v nízké koncentraci, koncentrace LH i FSH je zvýšená. Retinovaná abdominální varlata by měla být z důvodu rizika malignity odstraněna, pokud se pacient cítí jako žena, je ve v příslušném věku vhodné zahájení substituce estrogeny, pokud jako muž, je možná chirurgická rekonstrukce penisu, implantace protéz varlat a zahájení substituce angrogeny.
II. Porucha účinku androgenů
Mutace LH receptoru
Inhibiční mutace LH receptoru vede k hypoplázii Leydigových buněk a deficitu testosteronu i DHT díky porušenému účinku hCG i LH.
Enzymová porucha
- mutace StAR (steroidogenic acute regulatory protein) a CYP11A1 (cholesterol side-chain cleavage enzyme) vede k poruše reprodukce androgenů nadledvinami i varlaty. Postižení jedinci 46, XY mají rychle vznikající solnou poruchu díky selhání nadledvin a ženský fenotyp (byly popsány i lehčí varianty s opožděným nástupem příznaků).
- deficit 3β-hydroxysteroid dehydrogenázy typu 2 vede také k adrenální insuficienci, ale akumulace DHEA má mírně androgenizující efekt vedoucí k nejednoznačnému genitálu nebo hypospádii. U části dětí se projevuje také solná porucha.
- mutace CYP17 s deficitem 17α-hydroxylázy vede k hypertenzi a hypokalémii díky retenci sodíku při vysoké koncentraci kortikosteronu a 11-deoxykortikosteronu a nedostatečné androgenizaci nebo vzniku ženského fenotypu díky nedostatku androgenů (varlata mohou zůstat retinovány v inguinálním kanále).
- defekt 17β-hydroxysteroid dehydrogenázy typu 3 vede ke snížené produkci testosteronu.
- defekt 5α reduktázy vede 3 vede ke snížené produkci DHT.
Poruchy účinku androgenů
Syndrom necitlivosti k androgenům – mutace androgenního receptoru (AR) vede k rezistenci k testosteronu a DHT s četností nejméně 1:100 tisíc jedinců a karyotypem 46, XY. Při úplné necitlivosti mají 46, XY jedinci ženský fenotyp, prsa jsou normálně vyvinuta díky aromatizaci testosteronu na estrogeny, vzhledem k normální produkci AMH/MIS mají krátkou vaginu, ale bez dělohy, minimální pubické a axilární ochlupení a ženskou identitu i sexuální chování. Občas ve vyskytují tříslené kýly díky retinovaným varlatům, diagnóza je většinou zjištěna díky primární amenorey v pubertě. Vzhledem k nízkému riziku malignity by měly být dívkám odstraněny gonády spolu se zahájením substituce estrogeny v pubertě, popřípadě gonády mohou být odstraněny až po ukončení puberty a dokončení vývoje prsu (gonády jsou zdrojem testosteronu, který se aromatizuje na estrogen). Užití vaginálních dilatátorů většinou postačuje k udržení sexuálního života. Při částečné necitlivosti (Reifensteinův syndrom) je částečně zachována funkce AR, která se projevuje penoskrotální hypospadií, nedokončením sestupu varlat a vznikem gynekomastie. Pacienti se většinou identifikují jako muži, někteří úspěšně vstoupí do puberty, která může být podpořena vysokými dávkami testosteronu (tento postup ale není podpořen studiemi), častá je azospermie. Těžší necitlivost vede k ženskému fenotypu, pouze s velkým klitorisem a fůzí lábií.
III. Ostatní poruchy postihující 46, XY jedince
Syndrom perzistujícího Müllerova vývodu – díky mutaci genu pro AMH/MIS nebo jeho receptoru (AMHR2) nedojde k regresi orgánů vycházejících z Müllerova vývodu. Fenotypově muži mají přítomnou dělohu.
Izolovaná hypospádie – incidence je cca 1:250, většinou idiopatická.
Kryptorchismus – unilaterální forma postihuje při narození cca 3 % chlapců, u většiny z nich dojde k dokončení setupu v pozdějí době. orchidopexe je ke zvážení, pokud nedojde k sestupu do 6 – 9 měsíců věku. Při bilaterálním kryptorchismu, který je vzácnější, bývá častěji spojen s deficitem gonadotropinů nebo DSD.
46, XX DSD
Při androgenizaci plodu s karyotypem 46, XX (dříve nazývaný ženský pseudohermafroditismus) je přítomna nadprodukce androgenů buď z přítomné tkáně varlat nebo častěji původem z nadledvin.
I. 46, XX testikulární nebo ovotestikulární DSD
Varlata mohou být u karyotypu 46, XX přítomna u různých mutací (např. translokace SRYl, duplikace SOX9, dále u defektu RSPO1 nebo SF1/NR5A1).
II. Nadměrná expozice androgenům
Deficit 21-hydroxylázy (kongenitální adrenální hyperplázie)
- Klasická forma – incidence je 1:10 – 15 tisíc a je nejčastější příčinou androgenizace plodů 46, XX. Dochází k bloku syntézy adrenálních gluko- a mineralokortikoidů. Nedostatek glukokortikoidů vede ke kompenzatornímu zvýšení ACTH a hyperplázii nadledvin s dalším zvýšením syntézy steroidních prekurzorů proximálně od enzymatického bloku (hromadění 17-hydroxyprogesteronu a jejich směřováním do syntézy androgenů. Jejich zvýšená syntéza vede k androgenizaci plodu v prvním trimestru a nejednoznačným genitálem po porodu se zvětšením klitorisu a fůzí labií u žen. U mužů nadbytečná produkce androgenu vede k předčasné pubertě.
se solnou poruchou – aktivita 21-hydroxylázy je 0 %. Při těžkém kombinovaném deficitu glukokortikidů a mineralokortikoidů vzniká solná porucha, která se manifestuje v prvních 5 – 21 dnech života. Projevuje se hyponatrémií, hyperkalémií, hypoglykémií,. hypovolémií a nízkou hladinou gluko- a mineralokortikoidů, při vzniku šoku je choroba potenciálně život ohrožující. Vzhledem k závažnosti je u novorozenců zaveden screening k detekfci zvýšení 17-hydroxyprogesteronu). - prostá virilizující forma – aktivita 21-hydroxylázy je 1 %. Solná porucha není přítomna, u chlapcu vzniká předčasná puberta, u dívek je virilizace zevního genitálu a celková androgenizace.
- Neklasická forma – aktivita 21-hydroxylázy je 20 – 50%. U chlapců je klinicky němá, u dívek je hirsutismus, poruchy cyklu a infertilitaaktivita.
Základem terapie je urgentní tekutinová resuscitace, i.v. hydrkortizon a korekce hypoglykémie. Po stabilizaci pacienta je zavedena dlouhodobá terapie:
- glukokortikoidy ke snížení ACTH a zamezení hyperplázie nadledvin, snížení androgenizace, předčasné puberty a předčasného zrání skeletu. Cílem terapie je podání co nejmenších dávek glukokortikoidů, ale takových, aby došlo k potlačení nadprodukce androgenů nadledvinami. Základem bývá hydrokortizon, ale noční terapie prednisolonem se jeví jako výhodnější ke komplexnější supresi produkce ACTH. V těhotenství je potřeba se vyvarovat podávání dexametazonu.v CAVE Je potřeba pamatovat na nutnost navýšení dávek při stresu nebo onemocnění a mít stále u sebe pohotovostní kit.
- mineralokortikoidy u některých pacientů s prostou androgenizací. Adekvátnost této substituce lze posoudit plazmatickou reninovou aktivitou (CAVE hodnota se mění s věkem). Děti do jednoho roku často potřebují navíc i substituci soli, u dospělých často potřeba mineralokortikoidů klesá. Jejich nadbytek se projevuje hypertenzí a hypokalémií.
- k monitoraci celkové suprese lze použít hladinu 17-hydroxyprogesteronu, nicméně testosteron a androstenidion se zdá jako méně flukuující v čase.
- u nejtěžších případů je indikována adrenalektomie s nutností doživotní hormonální substituce.
- u dívek bývá často potřeba vaginální rekonstrukce a redukce velikosti klitorisu se zachování jeho nerového zásobení. Dále je potřeba pravidelná dilatace neovaginy a komplexní psychologická podpora. Dívky mají často sníženou fertilitu a trpí více syndromem polycystických ovárií.
- u mužů bývá hlavním zdrojem tvorby androgenů nadledviny, varlata bývají proto utlumena s rizikem snížení plodnosti.
Ostatní – zvýšení syntézy androgenů se projevuje i u deficitu:
- 11β-hydroxylázy (CYP11B1)
- 3β-hydroxysteroid dehydrogenázy (HSD3B2)
- aromatázy (CYP19)