Léčba lymfomů je vysoce specializovaná a následující přehled je pouze orientační…
Epidemiologie
- Incidence maligních non-Hodgkinských lymfomů (NHL) je cca 15/100000 obyvatel, Hodgkinova lymfomu (HL) 3/100000 obyvatel (v roce 2011 bylo v ČR diagnostikováno 2500 nových případů).
- NHL je chorobou staršího věku (medián 60 – 65 let) s některými výjimkami (lymfoblastový lymfom, primární mediastinální velkobuněčný lymfom).
- Převaha mezi pohlavím je u jednotlivých typů lymfomů individuální.
- Nejčastější je velkobuněčný lymfom z B lymfocytů (DLBCL, > 40%) a folikulární lymfom (FL, cca 20 %).
Etiologie
- Přítomnost lymfomu u příbuzných prvního stupně znamená zvýšení rizika vzniku lymfoproliferace 2 – 3x.
- Základním rizikovým faktorem je imunosuprese. Incidence lymfomů u HIV je cca dvacetinásobná (lymfoblastický lymfom až 500x vyšší) ve srovnání se zdravou populací. Podobně je i vyšší incidence u pacientů po transplantacích solidních orgánů nebo kostní dřeně s výrazným spolupodílem koinfekce EBV.
- Vyšší riziko lymfomů bývá při autoimunitních chorobách (tyreoiditida, Sjögrenův syndrom, celiakie apod.).
- Infekce řadou agens je spojena s rozvojem lymfomu, u některých je toto až patognomonické:
- Lidský T-lymfotropní virus 1 (HTLV 1) je spojen se vznikem T leukémie/lymfomu dospělých.
- Virus hepatitidy C zvyšuje riziko lymfoplazmocytárního lymfomu spojeného s kryoglobulinémií II. typu.
- U herpes viru 8 (HHV-8) je vyšší incidence primárního lymfomu charakteru výpotků a multicentrická Castlemanova choroba.
- Helicobacter pylorí má vztah k MALTomu žaludku,
- Borrelia burgdorferik MALTomu kůže,
- Ch. psittaci, Ch.pneumoniae a Ch. trachomatis k MALTomu spojivek,
- Campylobacter jejuni k MALTomu střev,
- EBV virus zvyšuje riziko Burkittova lymfomu, posttransplantační lymfoproliferace, DLBCL CNS, Hodgkinova lymfomu, extranodálního NL/T buněčného lymfomu nazálního typu.
- Zvýšená expozice herbicidů (dioxin) a pesticidů zvyšuje riziko B-NHL. Celkově riziko malignit zvyšuje expozice radiace
- Kouření zvyšuje riziko HL 2x.
…(tedy genetická zátěž, imunosuprese, zvýšená antigenní zátěž, autoimunita, infekce, radiace, kouření).
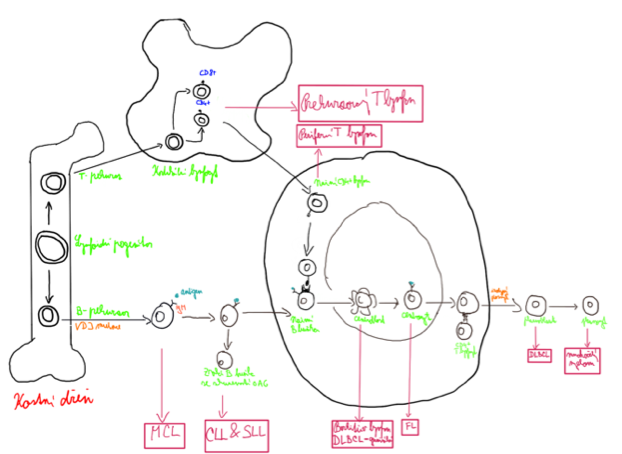
Patofyziologie – nádory vznikají na různé úrovni diferenciace lymfatického prekurzoru nebo buňky a v různých lymfatických orgánech (primárních kostní dřeň a thymus nebo sekundární mízní uzliny, slezina, MALT). Bývají často spojeny s translokacemi, které u B-NHL často postihují 14. chromozóm (zde jsou těžké řetězce imunoglobulinů) nebo translokace protoonkogenů s jejich deregulací:
- t(14;18) – 90 % FL, velká část DLBCL
- t(11;14) – MCL
- t(8;14) – Burkittův lymfom
- t(2;5) – ALCL
- postižení 14q32 – 70 % případů mnohočetného myelomu
Klinický obraz
Lymfomy postihují nejčastěji lymfatické uzliny, ale mohou postihnout jakýkoliv orgán a příznaky mohou být rozmanité. U části nemocných je náhodný nález zvětšených uzlin, u části vede k vyšetřování abnormální laboratorní nález v krevním obraze (anémie, trombocytóza nebo lymfocytóza při leukemizaci) + další specifické abnormality vyplývající z postižení konkrétního orgánu (alterace jaterních testů při postižení jater, hyperazotémie při postižení ledvin nebo útlakem ureterů tumorózní masou). Někdy se projevují dušností a kašlem při postižení mediastina a hilů s útlakem dýchacích cest, pleurálním a perikardiálním výpotkem nebo syndromem horní duté žíly. Při postižení uzlin v břichu může vzniknout břišní dyskomfort, imitace vředové choroby (zejména MALTomy žaludku). Postižení ilických a inquinálních uzlin mohou vést k otokům dolních končetin. Mezi celkové příznaky pak patří „B příznaky“ (horečky > 38°C, váhový úbytek > 10 kg/6 měsíců, noční poty). V podstatě na lymfom je potřeba myslet téměř u každého příznaku.
Klasifikace – klasifikace lymfoidních neoplázií se vyvíjela postupně během 20. století. Dle guidelines České hematologické společnosti je v současnosti doporučeno používat klasifikaci WHO (2016). Lymfomy je možné dělit na HL a NHL. Velmi složitá (on-line k dispozici zde: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4874220/).
NHL lze rozlišit na B-NHL (CD20, CD19, CD79) v 90 – 95 % a T-NHL (CD3) v 5 – 10 %. Dále se rozdělují na prekurzorové a periferní. Dle přežití 5 let lze NHL dále dělit na:
- indolentní – 5 let přežití > 70 % (FL)
- intermediální – 5 let přežití 50 – 70 % (DLBCL)
- agresivní – 5 let přežití < 50 % (MCL)
Diagnostika – vždy je zapotřebí velmi podrobné fyzikální vyšetření zaměřené na místa nejčastějších lokalizací (uzliny, játra, slezinu), dále CT vyšetření v místech nedostupných fyzikálnímu vyšetření (mediastinu, hily, ostatní uzliny hrudníku, břicha, ilik a inquin, dále slezina a játra). Poslední dobou se s úspěchem užívá PET CT (lymfomy avidní při PET CT jsou FL, DLBCL, MCL, HL a další). K potvrzení diagnózy je nutné histologické a imunohistologické vyšetření tkáně, nejčastěji uzliny, kdy je potřeba odebrat dostatečné a reprezentativní množství materiálu (volbou je získání zvětšené periferní uzliny, v případě nedostupnost thorakoskopie nebo jiná „skopie“ nebo „tomie“, pokud toto nedovolí stav pacienta volíme biopsii navigovanou pod CT nebo UZV). Důležité je i vyšetření kostní dřeně s event. imunofenotypizací, cytogenetikou a molekulárně biologickými metodami. Další vyšetření jsou indikována při podezření na speciální lokalizaci lymfomu (gastrofibroskopie, endosonografie, jaterní biospie, lumbální punkce event. MRI mozku).
CAVE V případě progrese choroby je nutná histologická revize (např. FL může progredovat do DLBCL).
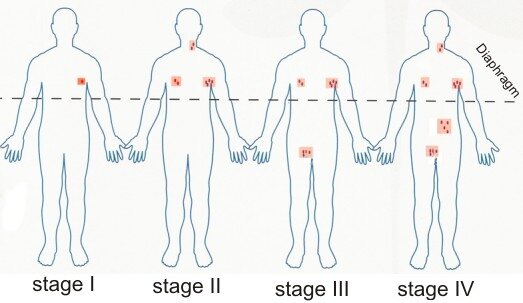
Staging – po potvrzení diagnózy je potřeba stanovit celkovou zdatnost nemocného (performance status 0-4) a stanovení rozsahu onemocnění (Ann Arbor klasifikace – tabulka 2).
Terapie – vždy je na chorobu potřeba pohlížet jako na potenciálně generalizovanou. Léčbu je třeba volit na základě biologické povahy nádoru (některé tumory jsou agresivně probíhající, ale potenciálně kurabilní, jiné benigní, pomalu progredující, ale téměř nevyléčitelné, kdy lze zpočátku zvolit strategii watch and wait). Způsoby léčby:
- Základní metodou je chemoterapie. Jde zejména o kombinovaný přístup založeném na kombinaci alkylační látka (cyklofosfamid) + antracyklin (adriamycin) + inhibitor mitózy (vincristin) + kortikoid (prednison) = CHOP (u méně agresivních tumoru lze vynechat antracyklin = COP). Při selhání se používají záchranné postupy založené na kombinaci s platinou (např. DHAP = cis-platina + cytosin arabinosid + metylprdnsolon nebo ICE = ifosfamid + karboplatina + etoposid).
- Vysokodávková terapie + autologní transplantace kostní dřeně znamená přístup: vyzabíjím všechny nádorové buňky i za cenu likvidace vlastní hematopoézy, kterou poté obnovím autologní transplantací. Věkový limit tohoto postupu je obvykle 65 – 70 let.
- Alogenní transplantace je použitelná pouze u některých typů lymfomů na principu graft v.s. lymphoma (GVL) efektu, kdy štěp zaútočí proti lymfomovým buňkám (většinou při selhání autologní transplantace). Velkou nevýhodou je, že štěp nelikviduje pouze lymfom, ale i ostatní buňky hosta tzv. graft v.s. host-disease (GVHD), která významně zhoršuje prognózu pacienta.
- Radioterapii lze využít izolovaně pouze v ojedinělých případech jednoznačného postižení pouze v jedné oblasti, jinak má své místo v kombinovaném postupu léčby bulky lymfomů.
- Imunoterapie je nyní jednoznačně na vzestupu. Principem je označení cílové buňky protilátkou s jejím následným zničením imunitním systémem pacienta nebo apoptózou. Základem léčby B lymfomů je anti-CD20 protilátka rituximab (často v kombinaci s chemoterapií = R-CHOP). Existuje nepřeberná řada dalších protilátek a nové stále vznikají.
- Podpůrná léčba je zásadním opatření a spočívá v aplikaci stimulujících faktorů (G-CSF, erytropoetin), substituci erytrocytů a trombocytů, podpora výživy, agresivní léčba infekcí, péče o vnitřní prostředí apod. V případě potřeby je vhodný odběr pohlavních buněk ještě před léčbou.
Odpověď na léčbu hodnocena následovně:
- CR – kompletní remise, kdy není lymfom zjistitelný
- PR – parciální remise s ústupem choroby > 50 %
- SD – setrvalé onemocnění s ústupem choroby < 50 %
- PD – progredující onemocnění
Prognóza – mezi znaky nepříznivé prognózy patří:
- horší stav nemocného (věk > 60 let, závažné komorbidity, PS III a IV)
- přítomnost B příznaků
- velikost tumoru (bulky postižení při velikost > 7,5 cm) a extralymfatické postižení
- vysoká hodnota β2-mikroglobulinu, LDH, anemizace, hypoalbuminémie aj.
Non-Hodgkinské lymfomy
Lymfomy prekurzorové
Lymfoblastový lymfom – tvoří s akutní lymfoblastickou leukémií jednu jednotku. V 85 – 90 % je z B buněk, zbytek je z T buněk. Často se šíří extranodálně včetně ALL. Je stejně agresivní jako ALL a podobá je i jeho léčba.
Lymfomy periferní z B-buněk
Difuzní velkobuněčný lymfom (DLBCL) – nejčastější lymfom (45 % všech lymfomů) vznikající z B-buněk (CD20+, CD3-). Věkový průměr je 60 let, u 50 % pacientů je extranodální postižení, u 15 % postižení kostní dřeně, u 50 % elevace LDH. Na základě genetického vyšetření se rozlišují dvě základní skupiny:
- GCB (vycházející z buněk zárodečného centra), nejčastěji s t(14;18) a mutací bcl-2
- non-GCB (vycházející z aktivovaných lymfocytů) s mutací bcl-6 a aktivací NFκB.
Základní léčbou je R-CHOP (u selektovaných skupin maxiCHOP a megaCHOP) event. vysokodávková chemoterapie a ASCT, při selhání záchranné režimy založené na platině. Pětileté přežití je 60 – 70 %. Zvláštním typem jsou:
- primárně mediastinální B-buněčný lymfom (MBCL), který je blízký HL a medián věku postižení je 35 let života.
- primárně velkobuněčný lymfom CNS, který se projevuje neurologickými příznaky. Průkaz je vhodný stereotaktickou biopsií (resekce je k ničemu) a léčí se chemoterapií.
Folikulární lymfom (FL) – druhý nejčastější lymfom (19 % všech lymfomů) vznikající z B-buněk (CD20 +, CD3-). Typická je přítomnost translokace t(14;18) pro gen bcl-2. Polovina nemocných má postiženou kostní dřeň, 1/3 vyšší hladinu LDH. Léčba je systémová chemoterapie (pouze u jednoznačně prokázané izolované lokalizace lze použít radioterapii). Přístup je přísně individuální od „watch and wait“ po systémovou chemoterapii (R-COP, R-CHOP, bendamustin…). Při dosažení remise je vhodná dlouhodobá léčba rituximabem k jejímu udržení. Vždy je nutná histologická konfirmace (vždy při progresi) pro riziko progrese do agresivnějšího tumoru (DLBCL).
Lymfom z plášťových buněk (MCL) – vzácnější CD20+ lymfom, iniciálně často pokročilý s extranodálním postižení, pro který je charakteristická translokace t(11;14). Muži jsou postiženi 3x častěji než ženy. Základem je chemoterapie s rituximabem, R-CHOP, R-DHAP, eventuálně vysokodávkovaná terapie spolu s ASCT, při relapsech ibrutinib, brexucaptagene autoleucel. Dlouhodobé přežití je cca 70 %. CD5+, CD19+, CD23-.
Lymfom z malých lymfocytů (SLL) – překrývá se s chronickou lymfocytární leukémií (CLL). Věkový medián 60 let, muži 1,5x častěji postiženy než ženy. Většinou je onemocnění v pokročilém stádiu, v případě leukemizace (lymfocyty > 5 ∙ 109) se již označuje jako CLL – pravděpodobně jde o jednu jednotku. Přežití je stejné jako u CLL (medián 7 – 8 let). Často je možné zvolit strategii „watch and wait“, pokud je pacient indikován k léčbě je volbou R-FC (rituximab + fludarabin + cykofosfamid). Imunofenotypizačně tedy CD5+, CD19+, CD23+.
Lymfoplazmocytární lymfom – věkový medián 60 let, muži mírně častěji postiženy než ženy. Velmi často současná nadprodukce IgM s příznaky hyperviskozity. Lymfoplazmocytární lymfom + nadprodukce IgM s hyperviskozitou = Waldenströmova makroglobulinémie. Při diagnóze je obvyklá generalizace do uzlin, kostní dřeně a sleziny. U části nemocných může přítomen kryoglobulin, který vede k Raynaudovu fenoménu, kloubním obtížím a periferní neuropatii. U asymptomatických nemoných není potřeba ihedn zahájení terapie. Jednou z možností je R-FC.
Lymfomy z marginální zóny (MALT lymfom)
- Extranodální lymfom z marginální zóny vzniká v místě přítomnostní lymfatické tkáně mimo uzliny. Zásadním patofyziologickým momentem je chronická antigení stimulace s postupnou klonální proliferací B-lymfocytů. V případě přerušení této stimulace je proces reverzibilní a po přeušení antigenní stimulace dochází k remisi. Při jejím dlouhém trvání může dojít k určitým mutacím a proliferace lymfatické tkáně se stává autonomní a nezávislá na antigenní stimulaci (tedy zde již eeadikace nepomůže). Dle lokalizace lze rozeznat žaludeční typ (chronická infekce Helicobacter pylori), spojivkový typ (chronická infekce Chlamydia psittaci), kožní typ (chronická infekce Borellia burgdorferi). Lymfom je sice primárně extranodální, ale mohou být postiženy i uzliny. Prognóza onemocnění je poměrně dobrá. V oblasti žaludku je hlavním typem léčby nejdříve snaha o eradikaci H. pylori, poté chemoterapie (chlorambucil, cyklofosfamid, rituximab) event. s radioterapií a až jako ultimum refugium gastrektomie.
- Splenický lymfom z marginální zóny je typický přítomností nádorových buněk v periferii a kostní dřeni a splenomegalií. Iniciálně je často volena splenektomie, event. chemoterapie jejíž součástí je rituximab. Prognóza onemocnění je relativně dobrá.
- Nodální lymfom z marginální zóny není častý. Má nejhorší prognózu ze všech MALT lymfomů. CAVE Vždy je nutné vyloučit extranodální lymfom z marginální zóny.
Burkittův lymfom – agresivní typ nádoru.
Lymfomy periferní z T-buněk
Nodální T-lymfomy
Anaplastický velkobuněčný lymfom (ALCL) – periferní T-lymfoproliferace u které rozeznáváme systémovou (CD30+) a kožní variantu (lokalizovaná, u které je vždy nutné vyloučit systémovou variantu. Typickou translokací je t(2;5) obsahující ALK gen, která se vyskytuje u části ALCL:
- ALK+ tvoří mladší pacienti a mají lepší prognózu (kurabilní u 75 % pacientů).
- ALK- tvoří starší pacienti, jsou často extranodální a mají horší prognózu (nutné agresivnější postupy).
Do léčebného algoritmu patří ASCT, aloSCT, nověji brentuzumab vedotin (anti-CD30)
Periferní T lymfomy blíže nezařaditelné (PTL NOS) – primárně nodální lymfomy s imunofenotypem zralých T buněk (CD3+, CD4+, CD8+), které nelze blíže zařadit, většinou s extranodálním postižením a nepříznivou prognózou. Věkový průměr nemocných je 60 let.
Vzácnější periferní T-lymfomy
- Angioimunoblastický T-buněčný lymfom se obvykle projevuje s generalizovanou lymfadenopatií a hepatosplenomegalií a často nepříznivým průběhem.
- T-buněčná leukémie/lymfom dospělých je agresivní lymfom spojený s HTLV-1 infekcí (nečastěji v Karibiku a východní Asii), často s osteolytickými ložisky.
Extranodální T-lymfomy
Kožní T-lymfomy
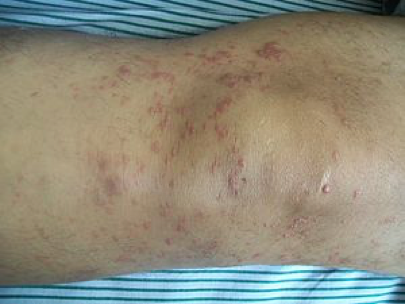
- Mycosis fungoides jsou má několik stadií. Zpočátku tvoří olupující se krusty nebo plochy s teleangiektáziemi a depigmetnacemi, postupně vředy až nádory se vkleslým povrchem připomínající klobouky hub (odtud název). V tomto stádiu bývají již postiženy i vnitřní orgány.
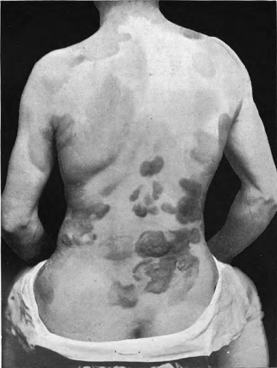
- Sézaryho syndrom se projevuje erytrodermií, generalizovanou lymfadenopatií s přítomností Sézaryho buněk a nepříznivou prognózou (medián přežití je 32 měsíců).
Obr. 2: Mycosis fungoides (vlevo a uprostřed), Sézaryho syndrom (vpravo)

- Primárně kožní CD30+ lymfomy
- Primárně kožní velkobuněčný lymfom CD30+se manifestuje lokálními uzly.
- Lymfomatoidní papulóza se projevuje noduly zejména na trupu a končetinách.
- Podkožní T-lymfom podobný panikulitidě se projevuje četnými podkožními uzly s dobrou prognózou.
Léčba kožních T-lymfomů je topická (kortikoidy, chemoterapie, retinoidy, fototerapie, radioterapie) nebo systémová (kortikoidy, imunoterapie, chemoterapie, fotoforéza)
Jiné než kožní T-lymfomy (často v Asii)
- NK/T lymfom nazálního typu je spojen s EBV infekcí, nejčastěji ve Střední a Jižní Americe a Asii. Jde o agresivní onemocnění projevující se infiltrací a destrukcí nosu paranazálních dutin a nosohltanu. Léčba je kombinací chemoterapií a radioterapií.
- T-lymfom spojený s enetropatií bývá často spojen s celiakií. Projevuje se bolestmi břicha, někdy s perforací střeva. Prognóza není optimální léčba je chirurgická (často vynucená komplikacemi) a systémovou chemoterapií.
- Hepatosplenický T-lymfom je agresivní onemocnění bez lymfadenopatie, ale s infiltrací kostní dřeně, jater a sleziny.
Hodgkinův lymfom
Epidemiologie
- Incidence je 3/100000/rok.
- Nejčastější věk výskytu je bifázický, první vrchol mezi 20 – 30 lety a poté po 50. roce života.
- Muži jsou mírně častěji postiženi než ženy.
Etiologie
- EBV infekce
- HIV infekce (100 % HIV pozitivních jsou zároveň i EBV pozitivní)
- Genetická predispozice (u sourozenců existuje 3 – 7x vyšší riziko vzniku HL ve srovnání s běžnou populací)
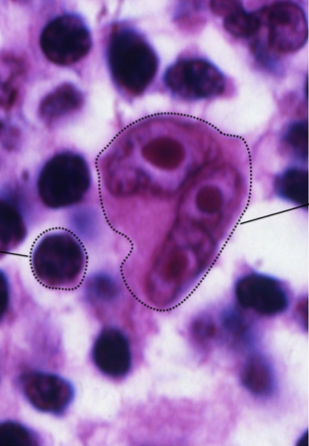
Patofyziologie – u nádorových buňkách byly prokázány IgVH mutace, které nastávají u B lymfocytů v sekundárním lymfatickém folikulu po jejich styku s antigenem, což svědčí pro B původ buněk (nenesou specifické znaky ani pro jednu lymfocytární řadu, na druhou stranu se předpokládá, že 1 – 2 % RS buněk může mít původ v T-lymfocytech). Hlavní příčinou je trvalá aktivace transkripčního faktoru NFκB a aktivace celé řady antiapoptotických mechanismů. Jedním z mechanismů je přítomnost EBV infekce vedoucí k nadprodukci LAMP1 (latentní membránový protein 1), který působí overexpresi NFκB. Diagnostickou je přítomnost RS buněk (Reese-Sternbergové).
CAVE RS buňky tvoří pouze 1 – 2 % nádorové populace, zbytek jsou lymfocyty a makrofágy.
Klinický obraz – nemocní typicky přichází se zvětšenými nebolestivými uzlinami (zejména krčními, axilárními, mediastinální a hilovými), většinou bez systémových příznaků. Někdy se HL projevuje útlakem zvětšenými uzlinami (kašel dušnost při zvětšení hilových a mediastinálních uzlin, někdy syndrom horní duté žíly). Někdy jsou přítomny B příznaky a choroba současně připomíná autoimunitní nebo systémovou chorobu.
Klasifikace (dle WHO)
- Nodulární HL s predominancí lymfocytů (CD30-, CD15-, CD20+)
- Klasický HL (CD30+, CD15+, CD20-)
- nodulárně sklerotický typ (70 %)
- smíšeně buněčný typ (20 – 25 %)
- na lymfocyty bohatý (5 %)
- s lymfocytární deplecí (1 %)
Diagnostika – základem je průkaz přítomnosti RS buněk nebo Hodgkinových buněk. Základními vyštřovacími metodami jsou RTG S+P, ultrazvuk, ale základem je CT a zejména PET CT.
CAVE I při nálezu hraničních uzlin (do 2 cm) mohou být přítomny B-příznaky.
K definitivnímu potvrzení je nutná biopsie uzlin se získáním dostatečně reprezentativnímu vzorku tkáně. Následně se provádí staging dle Ann-Arbor klasifikace.
Laboratorně bývá vyšší FW, dále leukocytóza, lymfopenie a anémie (negativně prognostické faktory) a dysfunkce T-lymfocytů při zachovalé funkci B-lymfocytů.
Terapie – HL je radio- i chemosenzitivní tumor, základním principem je systémová chemoterapie doplněná lokální radioterapií.
- Lokalizované onemocnění (Ann-Arbor I, II) bez rizikových faktorů se léčí ABVD (adriamycin, bleomycin, vinblastin, dakarbazin) + radioterapie. V přítomnosti rizikových faktorů u mladších nemocných se podává BEACOPP (bleomycin, etoposid, adriamycin, cyklosfosfamid, vinkristin, prokarbazin, prednison), u starších ABVD + radioterapie.
- Pokročilé formy (Ann-Arbor III, IV) onemocnění se léčí BEACOPP popř. ABVD v eskalovaných dávkách.
- Při relapsu se volí záchranné režimy založené na platině (DHAP (cisplatina, metylprendisolon, cytosin-arabinosid), ICE (ifosfamid, karboplatina, etoposid).
- Při selhání ASCT.
Prognóza – léčba je úspěšná u 95 % pacientů s lokalizovaným a 80 % s pokročilým onemocněním.