Definice – monoklonální nádorové choroby vznikající z pozdních fází vývojové řady B-lymfocytů. Za normálních okolností je jejich maturace do plazmocytů stimulována vazbou antigenu na jejich povrchový imunoglobulin. Při monoklonálních gamapatiích dochází k deregulaci jejich proliferace a nadměrné produkci imunoglobulinů.
V molekulách imunoglobulinů lze rozeznat tři úrovně odlišností, které působí jejich unikátnost:
- izotypy – existuje pět izotypů těžkých řetězců (M, A, G, D, E) a dva lehkých řetězců (κ, λ). Dávají vznik jednotlivým třídám imunoglobulinů.
- allotypy – malé rozdíly v imunoglobulinech stejné třídy).
- idiotypy – jsou unikátní u imunoglobulinů produkovaných jednotlivými klony plazmocytů.
Struktura protilátky – protilátky jsou složeny z dvou těžkých (cca 50 kDa) a dvou lehkých řetězců (cca 25 kDa). Každý řetězec je složen z konstantní domény (malá variabilita aminokyselin) a variabilní domény (výrazná variabilita aminokyselin). Lehké i těžké řetězce jsou navzájem spojeny pomocí disulfidových vazeb, tak, že variabilní domény všech řetězců navzájem sousedí. Variabilní regiony jsou vytvořeny přeskupením genů (VDJ u těžkých řetězců, VJ u lehkých řetězců) a umožňují tak specificitu imunoglobulinu pro daný antigen. Po expozici antigenu se může lehký řetězec kombinovat s jiným izotypem těžkého řetězce se vznikem izotypového přesmyku (to k jakému přesmyku dojde řídí cytokinové prostředí).
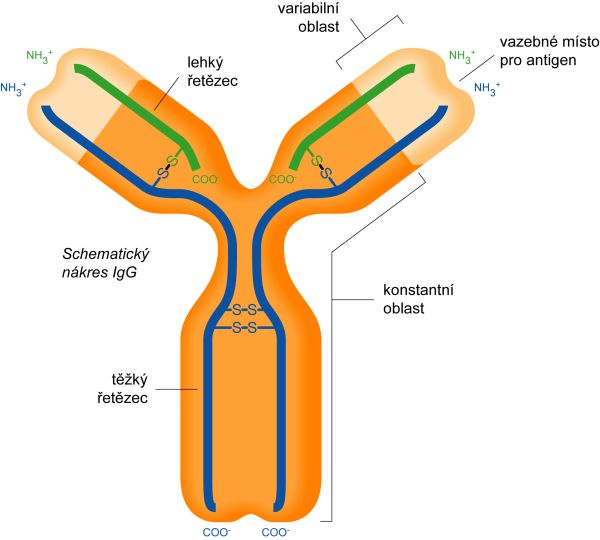
Ve většině případů jsou lehké řetězce produkovány v mírném nadbytku, které jsou jako volné lehké řetězce vylučovány ledvinami (normálně < 10 mg/den).
Imunoglobuliny se v elektrickém poli charakteristicky rozmisťují. Pokud je zde přítomen jasný vrchol (spike), je to známkou monoklonální tvorby protilátky typické pro danou distribuci a označuje se jako M komponenta (monoklonální). Tato M komponenta může být tvořena protilátkou nebo volným lehkým nebo těžkým řetězcem. Senzitivita této metody je >5 g monoklonální protilátky na litr. K potvrzení monoklonálního charakteru se doplňuje imunoelektroforéza, která typizuje daný typ volného řetězce.
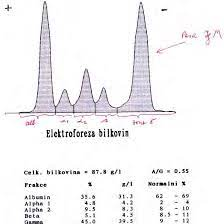
Množství M-komponenty je přímo úměrné náloži nádorových buněk. CAVE Monoklonální imunoglobulin je možné detekovat u celé řady dalších onemocnění (CLL, CML, lymfomy, karcinom prsu, tlustého střeva, jaterní cirhóza, sarkoidóza, parazitické choroby, pyoderma gangrenosum, pacienti po transplantacích apod). Dále se vyskytuje u dvou vzácných kožních chorob:
- lichen myxedematosus – vysoce kationické monokonální IgG se vychytávají v dermis.
- nekrobiotický xantogranulom – infiltrace kůže histiocyty (nejčastěji v obličeji) s tvorbou červených nebo žlutých uzlů. Přibližně u 10 % pacientů progreduje do myelomu.
Monoklonální gamapatie nejasného významu
Definice – monoklonální gamapatie nejasného významu (MGUS) je definována jako stav:
- přítomnosti monoklonálního imunoglobulinu do 30 g/l v krvi.
- < 10 % plazmatických buněk v kostní dřeni.
- nepřítomnost příznaků orgánového poškození (CRAB, hyperCalcemia, Renální insuficience, Anémie, Bone lesions).
Rizikový je vyšší věk (u 4 % > 50 let, 10 % > 80 let), častěji jsou postiženi muži, při postižení příbuzného prvního stupně MGUS/MM (OR 2 – 3), pracovníci s pesticidy a pacienti s autoimunitní chorobou. Existuje riziko přechodu do mnohočetného myelomu 1 % ročně (zvýšené při M komponentě non – IgG a/nebo její koncentraci > 15g/l nebo abnormálním poměru kappa/lambda řetězců). Stav se nijak neléčí, pouze vyžaduje observaci.
Mnohočetný myelom
Etiologie – maligní monoklonální proliferace plazmatických buněk, u které není zcela jasná etiologie. Roli hraje genetika (až 5x vyšší riziko při postižení příbuzných prvního stupně, četnější jsou různé trizomie chromozómů a translokace zahrnující 14. chromozóm, mutace N-ras, K-ras a B-raf byly kombinovaně zaznamenány u > 40 % pacientů, nicméně dosud nebyla identifikována kauzální mutace, která by byla prokázána alespoň u 20 % pacientů), dále je možný vliv ionizujícího záření a některých chemikálií (pesticidy, petrolej). Roli může hrát i IL-6.
Epidemiologie – incidence 1:25 tisíc, častěji ve vyšším věku (medián 69 let, raritní ve věku < 40 let), muži jsou postiženi 1,5x častěji než ženy, černí 2x častěji než bílí.

Klinický obraz a patofyziologie jeho vzniku – myelomové buňky se vážou na buňky stromatu kostní dřeně a na extracelulární matrix, což spouští jejich růst a proliferaci (mimo této adheze se spolupodílí i cytokiny, např. IL-6, IGF-1, VEGF, stromální růstový faktor SDF-1, dále proteinkinázy stimulované RAS/RAF a celkově plazmatické dendritické buňky a Th17 lymfocyty). Myelomové buňky interagují hlavně s osteoklasty a endotelovými buňkami. Hlavním příznaky lze vyjádřit eponymem CRABI:
- C (hypercalcemia) jako následek osteolýzy.
- R (renal) – k postižení ledvin dochází > 25 % pacientů. Nejčastější příčinou je hyperkalcémie a tubulární postižení precipitovanými lehkými řetězci, které se úvodem manifestuje Fanconiho syndromem (proximální renální tubulární acidóza typu II, glykosurie a aminoacidurie) a poruchou koncentrační schopnosti ledvin, nicméně uplatňuje se i další řada faktorů (recidivující infekce, užití NSAID, infiltrace glomerulů i celkově ledvin amyloidem). Jelikož jsou glomeruly většinou ušetřeny, bývá pouze minimální albuminurie. M komponenta je kationická, dochází k retenci Cl– jako kompenzace a bývá proto snížený anion gap. Pacienti jsou velice náchylní k renálnímu selhání při dehydrataci.
- A (anemia) – u cca 80 % pacientů se objevuje normocytární, normochromní anémie (většinou důsledkem útlaku hematopoézy v kostní dřeni plazmocyty, sníženou produkcí erytropoetinu selhávajícími ledvinami, její inhibicí produkty nádorových buněk a efektem terapie myelomu). Abnormality koagulace bývá způsobena poruchou destiček a interakcí faktorů I, II, V, VII a VIII s M komponentou (vzácně může vznikat „příznak mývala“, jehož nejčastější příčinou bývá jinak trauma). Pokud se ta navíc podílí na vzniku kryoglobulinů nebo vzniká hyperviskózní syndrom, je běžný Raynaudův fenomén. Běžná viskozita séra je 1,8 (ve srovnání s vodou), při hodnotách > 4 (hodnota paraproteinu > 40 g/l u IgM, > 50 g/l u IgG3 a > 70 g/l u IgA) se již projevuje hyperviskózní syndrom (typická trias – slizniční krvácení, neurologické symptomy (bolesti hlavy, poruchy vědomí až synkopy, parestézie a hypestézie) a změny vizu).

- B (bones) – bolesti kostí postihují > 70 % pacientů s myelomem (trvající bolest může znamenat patologickou frakturu). Myelomové buňky interagují hlavně s osteoklasty a endotelovými buňkami, aktivují OAF (osteoklast aktivující faktor) a působí osteolytické léze (bez významnější osteoprodukce) s mobilizací kalcia a vznikem hyperkalcémie, dále mohou vést ke vzniku kompresních fraktur obratlů se všemi neurologickými důsledky. Neuropatie spojené s myelomem bývají spíše senzorické, než motorické a častěji pokud je M komponentou IgM.
- I (infection) – je predispozice ke vzniku infekcí (zejména pneumonie – nejčastěji pneumokok, zlatý stafylokok a KLPN, dále pyelonefritidy – nejčastěji E. coli). Příčinou je difuzní hypogamaglobulinémie (mimo M komponentu) s potlačením produkce nemonoklonálních imunoglobulinů a jejich velice špatnou odezvou na antigenní stimulus (zejména polysacharidové složky buněčných stěn bakterií). Dalším faktorem je snížení Th1 odpovědi, zvýšení Th17 odpovědí, pomalejší migrace granulocytů a abnormality komplementu. Tuto situaci dále zhoršuje standardní terapie myelomu (např. dexametazon).
Klasifikace – vždy je třeba rozlišit jednotlivé jednotky:
MGUS – monoklonální protein v séru < 30 g/l + monoklonální plazmocyty v kostní dření < 10 % + absence orgánově specifických poškození odpovídajících mnohočetnému myelomu nebo AL – amyloidózy.
Doutnající myelom („smoldering myeloma“) – monoklonální protein > 30g/l v séru a/nebo > 0,5g/24h v moči (Bence – Jonesova bílkovina) a/nebo monoklonální plazmocyty v kostní dření v množství 10 – 60 % všech buněk + současná absence orgánově specifických poškození odpovídajících mnohočetnému myelomu nebo AL – amyloidózy. Při přítomnosti popsaných abnormalit (paraprotein > 30g/l + > 0,5g/24h v moči + plazmocytóza dřeně) je 5 leté riziko progrese do mnohočetného myelomu 76 %.
Mnohočetný myelom – kritéria:
- I. Monoklonální infiltrace kostní dřeně (> 10 % plazmocytů jako malé kritérium nebo > 30 % plazmocytů jako velké kritérium) a/nebo extramedulární plazmocytom
- II. Jeden nebo více z následujících příznaků postižení cílových orgánů – CRAB:
- (C – calcium) – kalcémie > 2,75 mmol/l
- (R – renal) – GF < 0,66 ml/s a/nebo Skreat > 177 μmol/l.
- (A – anemia) – anemie, hemoglobin pod 100 g/l nebo 20 g/l pod dolní limit normy
- (B – bone) – osteolytické kostní destrukce nebo osteoporóza
- III. Monoklonální komponenta (IgG > 35 g/l nebo IgA > 20 g/l nebo množství lehkých řetězců v moči za 24 hodin > 1 g).
Nesekreční myelom – splněna pouze kritéria I. + II. (není monoklonální komponenta v krvi).
Solitární kostní plazmocytom – solitární lytická kostní léze bez infiltrace kostní dřeně. Citlivý k radioterapii. Má dobrou prognózu.
Extramedulární plazmocytom – infiltrace submukózní lymfoidní tkáně nosohltanu nebo paranasálních dutin bez infiltrace kostní dřeně. Citlivý k radioterapii. Má dobrou prognózu.
Plazmocelulární leukémie – plazmatické buňky v periferní krvi > 2 × 109 /l nebo > 20 % z celkového počtu leukocytů.
Diagnostika
- 1. Zásadním je stanovit přítomnost, typ a množství monoklonálního paraproteinu v krvi popř. moči (Bence-Jonesova bílkovina) pomocí elektroforézy a imunofixace. Stanovit lze i koncentraci samotných volných lehkých řetězců v séru. Paraprotein ale může chybět ve vzácných případech nesekretorického myelomu. Četnost jednotlivých typů imunoglobulinů: IgG v 53 %, IgA ve 25 %, IgD v 1 %, volné lehké řetězce ve 20 %. U IgM paraproteinu je výrazně častější hyperviskózní syndrom.
- 2. Charakteristickým nálezem je velmi vysoká sedimentace erytrocytů (FW).
- 3. Při nálezu > 2 . 109/l plazmocytů v krvi jde již o plazmocytární leukémii (přítomna výrazně častěji u IgD a IgE typu).
- 4. I přes osterolýzu bývá ALP normální, protože není téměř přítomna osteoblastická aktivita. Důležitá je i hladina β2-mikroglobulinu a albuminu (ISS staging).
- 5. RTG hrudníku, lebky a dlouhých kostí může odhalit osteolýzu nebo difuzní osteopenii. Moderním přístupem je průkaz osteolýz pomocí low-dose celotělového CT. Podobně lze prokázat zvýšený metabolismus v místě mnohočetného myelomu/plazmocytomu pomocí 18F-FDG PET/CT.
Staging – při myelomu je nejčastějším typem imunoglobulinu IgG (více než 50 %), IgA (více než 20 %), volné lehké řetězce (20 %) a zbývajících cca 1 % ostatní typy. Spíše historicky se uvádí klasifikace dle Durieho a Solomona:
- Klinická stádia
- I. stádium – IgG < 50 g/l, IgA < 30 g/l, v moči < 4 g/24h, přítomno žádné nebo maximálně jedno osteolytické ložisko, nejsou ostatní příznaky (kalcium i hemoglobin jsou v normě).
- II. stádium – hodnoty mezi I. a III. stádiem
- III. stádium – IgG > 70 g/l, IgA > 50 g/l, v moči > 12 g/24h, přítomno mnohočetné ložiskové postižení, hyperkalcémie > 2,75 mmol/l, hemoglobin < 85 g/l.
- …tedy IgG – 50 – 70 – g/l, IgA – 30 – 50 g/l – g/l, protein v moči – 4 – 12 – g/24h.
- Dle renálních parametrů – při hodnotě kreatininu < 177 umol/l – A, při hodnotě > 177 umol/l – B
V současnosti se používá mezinárodní prognostický systém ISS, který je založen na hodnotách beta2-mikroglobulin (b2m) a albuminu (alb):
- Stadium I – b2m < 3,5 mg/l, alb > 35 g/l
- Stádium II – b2m 3,5 – 5,5 mg/l nebo b2m < 3,5 mg/l + albumin < 35 g/l
- Stádium III – b2m > 5,5 mg/l
Revidované ISS (R-ISS) používá navíc i hladinu laktát dehydrogenázy (online zde: https://www.mdcalc.com/revised-multiple-myeloma-international-staging-system-r-iss).
Prognóza – nejsilnějším prediktorem prognózy je hladina sérového β2-mikroglobulinu (lehký řetězec hlavního histokompatibilního antigenu I. třídy (HLA-A, -B, -C) na povrchu každé buňky). Prognózu ovlivňuje i přítomnost mutace (KRAS. NRAS, TP53, DIS3, FAM46C a BRAF), karyotyp, genotyp, hladina LDH. Vzhledem k novým možnostem léčby se přežití neustále prodlužuje.
Terapie
MGUS – žádná specifická terapie není indikována. Vhodné jsou kontroly á 1 rok (v případě vysokorizikového MGUS á 6 měsíců) k včasnému zachycení transformace do mnohočetného myelomu.
Doutnající myelom – observace se zahájením terapie v přítomnosti příznaků („CRAB“).
Mnohočetný myelom – úplné vyléčení pomocí vysokodávkové terapie a transplantace autologní kostní dřeně je 10 – 40 %. Léčba by vždy měla být zahájena v případě vzniku symptomů („CRAB“). Základem je zahájení vstupní indukční fáze (thalidomid/lenalidomib/bortezomib), poté myeloablace (většinou vysokodávkovaný melfalan), poté autologní transplantace kostní dřeně, poté konsolidace (obvykle jiný lék než indukce) a udržovací terapie lenalidomidem.
U seniorů lze použít podobný postup pouze bez myeloablace a transplantace. Režim je nutné volit více individuálně, doporučený je bortezomib-lenalidomid-dexametazon.
Nejdůležitějším pokrokem poslední doby je použití nových léčebných postupů (zejména při relapsu) a z nich hlavně antiCD38 (darutumumab). Základem je inhibitor proteazomu (bortezomib) nebo imunomodulační látka (thalidomid, lenalidomid) + dexametazon + nový lék do trojkombinace (karfilzomib, ixazomib, daratumumab, elotuzumab). Při refrakteritě k lenalidomidu se užívá kombinace pomalidomid + dexametazon + bortezomib/elotuzumab/antiCD38 monoklonální protilátky.
Látky:
- inhibitory proteazomu (bortezomib, karfilzomib, ixazomib)
- imunomodulační léky (thalidomid, lenalidomid, pomalidomid)
- monoklonální protilátky (daratumumab, isatuximab, elotuzumab)
- ostatní (panobinostat, selinexor).
Radioterapie se používá na bolestivá kostní ložiska spolu s bisfosfonáty. U hyperviskózního syndromu je indikována plazmaferéza, u hyperkalcémie a akutního renálního selhání dialýza popř. speciální modifikace s high-cutoff membránou, která odstraňuje lehké volné řetězce. Zejména při použití imidů je zvýšené riziko trombózy (při vysokém riziku je profylaxe pomocí LMWH, u nízkého rizika antiagregace). Při anémii je ke zvážení podpor pomocí erytropoetinu.
Waldenströmova makroglobulinémie
Historie – v roce 1948 popsal Waldenström maligní onemocnění z lymfoplazmocytárních buněk, které produkují IgM. Na rozdíl od myelomu zde dominuje lymfadenopatie, hepatosplenomegalie a zejména hyperviskózní syndrom.
Epidemiologie – medián věku odhalení choroby je 64 let, mírně častěji u mužů.
Patogeneze – nádorové buňky pochází z paměťových B – buněk nesoucích IgM, které již prošly somatickou mutací a antigenní selekcí v lymfoidním folikulu. Pacienti s IgM myelomem a Waldenströmovou makroglobulinémií (WM) se liší klinickým obrazem (u IgM myelomu jsou přítomny osteolytické léze a kostní dřeň je infiltrována CD138+ plazmocyty). Bylo identifikováno množství genetických mutací spojených se zvýšeným rizikem WD, ale žádná z nich nebyla kauzální.
U některých pacientů může být IgM namířen proti MAG (myelin-associated glycoprotein) a může být příčinou demyelinizace a periferní neuropatie pozorované u některých pacientů s WM.
V kostní dření se nachází > 10 % lymfoplazmocytárních buněk (pozitivita IgM+, CD19+, CD20+ a CD22+ – typické pro B – lymfocyty, negativita CD10− a CD23−) a zvýšený počet mastocytů. M – komponenta je > 30 g/l, ale na rozdíl od myelomu je pouze malé množství vylučováno močí (renální postižení nebývá přítomno).
Klinický obraz – dominuje lymfadenopatie, hepatosplenomegalie a zejména hyperviskózní syndrom (typická trias – slizniční krvácení, neurologické symptomy (bolesti hlavy, poruchy vědomí až synkopy, parestézie a hypestézie) a změny vizu se segmentací a dilatací cév), nejsou přítomny osteolytické léze, hyperkalcémie ani postižení ledvin. Častější je i řetízkovaní erytrocytů a pozitivní Coombsův test. 10 % makroglobulinů jsou kryoglobuliny s možností vzniku Raynaudova fenoménu.
Diagnostika WM = lymfoplazmocytární infiltrát v kostní dření při trepanobiopsii (!!! – nikoliv aspirací kostní dřeně) + IgM paraprotein.
Terapie – většinou nebývá potřeba, pokud nejsou přítomny příznaky (anémie, hyperviskózní syndrom, lymfadenopatie nebo hepatosplenomegalie:
- Plazmaferéra – indikována při výrazných příznacích hyperviskózního syndromu, protože 80 % paraproteinu je intravaskulárně.
- Chemoterapie – léčbou první volby jsou ibrutinib (inhibitory Brutonovy tyrozinkinázy) + alkylační látky (bendamustin, cyklofosfamid) + inhibitory proteazomu (bortezomib, carfilzomib, ixazomib) event. v kombinaci s rituximabem. Léčba relapsů již přesahuje rámec tohoto sdělení.
POEMS syndrom
Patogeneze – nejasná, byly prokázány vysoké hodnoty prozánětlivých cytokinů (IL-1, -6, VEGF a TNF-α).
Klinický obraz – je přítomna Polyneuropatie, Organomegalie, Endokrinopatie, M-komponenta a kožní změny (Skin changes). Celkově klinickému obrazu dominuje závažná polyneuropatie.
- Polyneuropatie – obvykle přítomna těžká senzomotorická polyneuropatie spojená s kostními sklerotickými změnami.
- Organomegalie – typická je hepatomegalie a lymfadenopatie, méně splenomegalie.
- Endokrinopatie – projevuje se jako hypogonadismus, méně jsou přítomny ostatní endokrinopatie (hyperprolaktinémie, hypotyreoidismus, adrenální insuficience).
- M komponenta
- kožní změny (Skin changes) – různě vyjádřené (hyperpigmentace, hypertrichóza, ztluštění kůže, paličkovité prsty, hemangiomy).

Terapie – podobná jako v případě mnohočetného myelomu, osteolytická ložiska lze léčit lokální radioterapií. Plazmaferéza nebývá účinná.