Úvod – funkce aminokyselin je rozmanitá. Slouží jako základní stavební kámen proteinů (enzymy, hormony…), neurotransmitery (glycin, glutamát, GABA). Osm aminokyselin (esenciální) si neumí organismus sám syntetizovat (valin, leucin, izoleucin, fenylalanin, tryptofan, lyzin, methionin a threonin). Názvosloví je dáno jménem hromadící se aminokyseliny + tekutina kde se hromadí (-emie v krvi, -urie v moči). Genotypová i fenotypová heterogenita je individuální u každé poruchy a je většinou výrazná. Diagnóza je většinou stanovena vyšetřením plazmy (iontová chromatografie), moči (plynová chromatografie, spektrometrie), stanovením acylkarnitinového profilu a potvrzena genetickým vyšetřením.Souhrnná prevalence poruch metabolismu aminokyselin je 1 : 1000 porodů a u některých chorob se provádí novorozenecký screening. Některé choroby ovšem mohou zůstat až dospělosti asymptomatické a mohou se prozradit pouze při hladovění nebo velkém stresu.
Fenylketonurie
Etiopatogeneze -AR dědičná porucha fenylalaninhydroxylázy (hydroxyluje fenylalanin za vzniku tyrozinu) s prevalencí 1 : 10 tisíc, pro kterou je charakteristické zvýšení fenylalaninu a jeho produktů v tělesných tekutinách. Důsledky:
- akumulace fenylalaninu inhibuje transport ostatních aminokyselin nutných k syntéze neurotransmiterů i myelinu s následnou nadměrnou produkcí noradrenalinu a serotoninu, což vede k těžké mentální retardaci.
- fenylalanin inhibuje syntézu melaninu, což vede k hypopigmentaci kůže a vlasů s tendencí k tvorbě ekzémů.
- akumulace fenylacetátu v moči a potu vede k jejich zatuchlému odéru (jako myšina).
Klinický obraz – postižené a neléčené děti jsou v době porodu zdravé, ale velmi rychle dochází k mikrocefalii a progresivní poruše funkcí mozku (demence, křeče, hyperaktivita, poruchy na EEG).
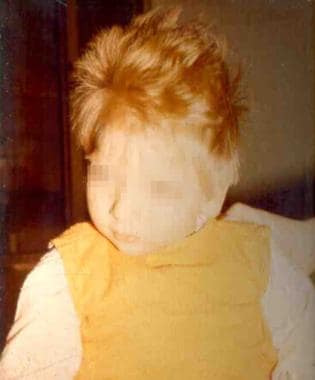
Diagnostika – k prevenci vzniku chronických změn je třeba zahájit léčbu do 2 týdnů věku dítěte. Z toho důvodu se v Evropě i USA provádí novorozenecký screening.
Terapie – základem léčby je specifická dieta, která:
- nesmí obsahovat fenylalanin
- musí být naopak bohatá na tyrosin, který se při dysfunkci fenylalanin hydroxylázy stává esenciální aminokyselinou
- pacienti s lehčí formou choroby mají lepší toleranci dietních prohřešků po suplementaci tetrahydrobiopterinem, který je kofaktorem fenylalanin hydroxylázy
CAVE Ženy, které důsledně dodržují režimová opatření, mohou otěhotnět. Před těhotenstvím i během něj je zásadním opatřením striktně dodržovat režimová opatření. Vzhledem k tomu, že vysoké hladiny fenylalaninu jsou teratogenní, je v opačném případě vysoké riziko vzniku mateřské fenylketonurie (vrozené vývojové vady, mikrocefalie a po narození těžká mentální a růstová retardace).
Homocysteinurie
Existuje osm různých druhů homocysteinurie, které vedou ke zvýšení koncentrace homocysteinu v moči a krvi.
Klasická homocysteinurie – nejčastější forma (četnost 1 : 200 tisíc) způsobená deficitem cystation-β-syntázy. U většiny pacientů se projevuje ve 3 – 5 letech věku dislokací oční čočky a mentální retardací (50 % případů), u některých je marfanoidní habitus s příznaky osteoporózy. Během první dekády dochází k život ohrožujícím vaskulárním komplikacím (postihující koronární, renální a mozkové tepny). Diagnózu lze prokázat zvýšením koncentrace metioninu, volného homocystinu (disulfidová forma) a plazmatického homocysteinu. Léčba:
- speciální dieta s restrikcí proteinů a metioninu a bohatá na cystin
- substituce pyridoxinem je účinná u cca 50 % pacientů, důležité je i doplnění folátu a vitamínu B12, pokud existuje deficit
Hyperhomocysteinémie – zvýšená hladina homocysteinu. Příčiny:
- heterozygoti a mírné formy homocysteinurie
- vysoký věk, kouření, postmenopauzální ženy, selhání ledvin, hypotyreoidismus, leukémie, IBD, psoriáza, léky (metotrexát, oxid dusný, isoniazid, antiepileptika).
Homocystein působí atero- a trombogenně a je nezávislým rizikovým faktorem kardiovaskulárních a trombotických komplikací. Substituce folátu a vitamínu B12 má efekt na redukci hladiny homocysteinu, ale bez vlivu na četnost kardiovaskulárních komplikací.
CAVE Hyperhomocysteinémie spolu s deficitem folátu a vitamínu B12 u těhotných žen je rizikovým faktorem vzniku defektu neurální trubice u jejich plodů.
Alkaptonurie
Vzácný (1 : 200 tisíc) AR dědičný deficit homogentisát 1,2-dioxygenázy, která se uplatňuje v metabolismu tyrosinu. Vede k akumulaci homogentisové kyseliny v moči a její oxidované formy v pojivové tkáni, kde působí pigmentaci (ochronóza). Může zůstat nerozpoznána až do středního věku. Příznaky:
- tmavá moč (u 1/2 pacientů)
- tmavé pigmentace (skléra, uši, kůže, sliznic a vnitřních orgánů) většinou vzniká ve 30 až 40 letech věku
- depozita v srdci a cévách vedou ke vzniku aortální stenózy, která si vyžádá operaci v cca 60 letech věku
- degenerativní postižení kloubů s omezením pohybu a bolestí (pánev, kolena, ramena). Akutní ataka může připomínat revmatoidní artritidu, ale malé klouby jsou spíše ušetřeny.

Diagnóza je dána zjištěním vysoké hladiny kyseliny homogentisové ve tkáních. Léčba:
- symptomatická (antiflogistika, ortopedická korekce kloubů, operace srdce)
- nízkoproteinová dieta s podáním nitisonu (léky na tyrozinémii I) vedou ke snížení hladiny kyseliny homogentisové v moči, což může vést k prevenci dlouhodobých komplikací
Defekty enzymů ureového cyklu
AR dědičné (deficit ornitin transkarbamylázy je X dědičný) poruchy enzymu ureového cyklu vedou ke vzniku nadbytku dusíku z proteinů, ze kterého vzniká nadbytek amoniaku a glutaminu vedoucí k neurotoxicitě a edému mozku. Souhrnná incidence je 1 : 25 000. Poruchy s klasickou poruchou se začínají projevovat po 1 – 4 dnech života, novorozenci odmítají jíst, postupně upadají do kómatu a umírají. Mírnější defekty se projevují odmítáním proteinů, opakovaným zvracením, střídáním nálad, podrážděností, chronickou únavou, dezorientací a progresí do kómatu. Diagnóze napovídá zvýšená plazmatická hladina amoniaku a aminokyselin. Léčba je zaměřena na přerušení katabolismu a produkce amoniaku:
- Důležitá je kalorická podpora (i.v. glukóza a mastné kyseliny), při potřebě inzulín.
- Nadbytek dusíku je vyvázán i.v. fenylacetátem (vyváže glutamin → fenylacetylglutamin) nebo benzoátem (vyváže glycin → hippurová kyselina). Oba finální metabolity jsou dobře rozpustné ve vodě a z těla odstraněny močí.
- Arginin se stává esenciální aminokyselinou (mimo deficit arginázy) a musí být substituován (200 mg/kg/den)
- Při selhání uvedených opatření je indikována hemodialýza.
Chronická terapie je založena dietě s restrikcí proteinů a podávání fenylbutyrátu, argininu (nebo citrulinu). V těžkých případech je ke zvážení transplantace jater.
Hyperamonémie při funkčním deficitu glutamin syntázy může vznikat po chemoterapii nebo imunosupresi po transplantaci orgánů, dále cirhóze jater. Léčba je podobná jako u vrozených defektů enzymů ureového cyklu.