Složení pojivových tkání – pojivové tkáně (kost, chrupavka, kůže, vazy a šlachy) jsou složeny z cca 500 různých substancí, které vytváří komplexní síť kolagenu, proteoglykanů, glykoproteinů a proteinů.
Nejběžnějšími substancemi jsou kolageny typu I, II a III, které mají pevnost ocelového lanka stejného kalibru. Kolagen I je nejčastější v dermis, šlachách, vazech a demineralizované kosti, II v chrupavce a I a III ve stěně velkých cév. Během dospělosti je obrat většiny tkáně velmi pomalý s výjimkou kosti, kde je středně intenzivní. Během určitých procesů převáží degradace nad syntézou:
- při hladovění je velká část kolagenu degradována, aby poskytl aminokyseliny ke glukoneogenezi
- pří artróze a revmatoidní artritidě dochází ke zvýšené degradaci chrupavky
- glukokortikoidy oslabují většinu tkání díky snížení produkce kolagenu
- při inaktivitě nebo při nízké gravitaci převáží degradace nad syntézou
Naopak u řady patologických stavů dochází k nadměrné fibroprodukci se vznikem vazivové náhrady poškozeného parenchymu (jaterní cirhóza, plicní fibróza, nefroskleróza, ateroskleróza).
Struktura a syntéza kolagenu
- Po syntéze α řetězce ribozomy (prokolagenové monomery – celkem cca 1000 aminokyselinových zbytků) jsou signální globulární peptidy na N-konci odštěpeny. V řetězci je každá třetí aminokyselina glycin, tedy opakují se sekvence -gly-X-Y- (X a Y jsou velice často prolin a hydroxyprolin). Prolinová rezidua na -Y- pozici jsou hydroxylovány pomocí enzymu prolylhydroxylázy na hydroxyprolin (esenciálním kofaktorem je vitamín C), což je zásadní pro konformaci do tvaru trojité šroubovice.
- Následně dochází k interakci a tvarování tří proα řetězců do trojité šroubovice, což je zahájeno na C-terminálním konci a odtud je šroubovice „pletena“ mechanismem podobným zipu směrem k N-terminálnímu konci. Po spletení všech řetězců dochází k odštěpení C- a N-terminálních propeptidů, což cca 1000x sníží rozpustnost ve vodě. Vzniklé fibrily jsou již samy o sobě pevné, ale jejich pevnost je dále zvýšena vznikem příčných můstku, kterými jsou jednotlivé fibrily spojeny. Kolagenová vlákna jsou rezistentní k většině proteáz a k jejich rozštěpení je potřeba speciální metaloproteináza kolagenázy, která vede k rozpletení trojšroubovice na gelatinózní strukturu, která je dále štěpena méně specifickými proteinázami. Existují i méně běžné typy kolagenů, které obsahují repetitivní sekvenci -gly-X-Y- (celkem 28 druhů). V elektronovém mikroskopu je patrno proužkování, které naznačuje precizní konformaci do fibril. Jednotlivé typy kolagenu řetězce se liší svým složením – kolagen I: α1(I)-α1(I)- α2(I), kolagen II: α(II)-α(II)- α(II), kolagen III: α(III)-α(III)- α(III).
Fibrilin a elastin – ve tkáních, které potřebují elasticitu (plíce, velké cévy, vazy), tuto elasticitu zajišťuje velký glykoprotein fibrilin, který obsahuje velké množství domén, podobných epidermálnímu růstovému faktoru, které jsou střídány doménami bohatými na cystein.
Proteoglykany – odolnost některých tkání vůči kompresi (aorta, chrupavka) je dána přítomností velkého množství proteoglykanů (identifikováno nejméně 30 druhů), které jsou složeny z kmenového proteinu, na který je přichyceno množství negativně nabitých disacharidů (často chondroitinsulfáty) a díky tomu vážou velké množství vody a malých iontů.
Osteogenesis imperfecta (OI)
Patofyziologie – za > 90 % všech případů OI je odpovědna heterozygotní mutace genů kódujících proα1 nebo proα2 řetězec prokolagenu typu I – COL1A1 nebo COL1A2.
Klinický obraz – typ II je nejzávažnější. Typická je:
- Zvýšená tendence ke vzniku zlomeniny. Během puberty se četnost zlomenin často snižuje, důležitá je zejména péče o ženy během těhotenství a po menopauze, kdy se jejich četnost opět zvyšuje.
- Oči bývají modré díky ztenčení kolagenové sítě a prosvítání choroidální vrstvy.
- Po 20 letech věku začíná docházet ke ztrátě sluchu (zhoršení osifikace středního ucha).
- Zuby mohou být různě postižené, mohou mít žlutou, šedou nebo namodralou barvu díky nedostatku dentinu (obsahuje mnoho kolagenu I). Zuby některých pacientů jsou značně fragilní a musí být extrahovány.
- Ostrůvky nepravidelné osifikace se strakatým vzorem na RTG lebky. Občas je přítomna hypermobilita kloubů a kardiovaskulární manifestace (aortální regurgitace, vlající mitrální chlopeň s regurgitací, fragilita velkých cév). Z neznámých příčin mají pacienti hypermetabolický stav se zvýšenou hladinou tyroxinu, hypertermií a excesivním pocením.



Klasifikace
- Typ I – Incidence je 1:15 – 20 tisíc. Tato forma může být limitující nebo oligo- nebo až asymptomatická. Většina pacientů má modré skléry.
- Typ II – nejzávažnější, incidence je 1:60 tisíc (velká část pacientů ale zemře in utero nebo časně po narození). Osifikace kostí není dokončena, pacient často trpí zlomeninami většiny kostí. Dle RTG lze rozlišit subtypy IIa, IIb, IIc.
- Typ III – vede ke středním až závažným deformitám kostí se vznikem deformit. Jsou normální skléry. Kyfoskolióza často omezuje dýchání.
- Typ IV – lehké až střední kostní postižení, může být podobné typu III, normální skléry.
- Typ V – lze rozeznat přítomností dislokovaných hlaviček rádií a tvorbou hyperplastických kalusů.
- Typ VI – XXI – geneticky definované
…I – mírná + modré, II – těžká + modré, III – střední až těžká + bílé, IV – střední + bílé
Klasifikace do jednotlivých typů nemusí ale nutně odrážet průběh onemocnění, který může být značně variabilní. Pro všechny OI je typické snížení kostní denzity. Kostní zlomeniny se ale hojí překvapivě normálně.
Diagnostika – k diagnóze většinou stačí klinický obraz, vždy je ale nutné vyloučit ostatní příčiny patologických fraktur (násilí, malnutrice, malignity, hypofosfatásie, chondrodysplázie). Nápomocná může být mikroskopie kostí.
Terapie – u některých pacientů není potřeba žádná terapie. Důležitá je opatrná fyzioterapie, léčba již vzniklých zlomenin a komplikací (neurologických komplikací, pneumonie apod.). V těžkých případech jsou indikovány bisfosfonáty. U závažných poruch sluchu lze pomoci náhradou třmínku. Zkouší se terapie kmenovými buňkami. U těžkých případů lze diagnostiku prenatálně provést pomocí ultrazvuku v 16 týdnu gravidity a rutinně vyšetřením DNA.
Ehlers – Danlosův syndrom (EDS)
Epidemiologie – incidence je 1 : 5000 živě narozených, mnohem častější u černé rasy. Nejčastější je hypermobilní forma.
Patofyziologie – za EDS je odpovědná mutace genu pro kolagen (druh postiženého genu se liší dle typu).
Klasifikace – zatím bylo prokázáno 13 subtypů EDS. Jednotlivé formy se mohou vzájemně překrývat.
- Typ I – těžká forma choroby s hypermobilitou kloubů a elasticitou kůže.
- Typ II – lehčí forma choroby s hypermobilitou kloubů a elasticitou kůže.
- Typ III – hypermobilita kloubů je výraznější než elasticita kůže.
- Typ IV (vaskulární)- výraznější je elasticita kůže (vznikají hyperpigmentace a jizvy nad kostními prominencemi), pacienti jsou ohroženi náhlou smrtí pro rupturu velkých cév.
- Typ V – podobný typu II, ale má X vázanou dědičnost.
- Typ VI (okulárně skoliotický typ) – fragilita očí s keratokonem + skolióza.
- Typ VII – podobný typu III.
- Typ VIII (periodontický) – dominují změny periodontu, kůže je spíše křehká a jizvící se, než elastická.
- Typ IX – XIII
…I – těžká, II – lehká, IV – postižení cév, VI – postižení očí s fragilními bulby.
Klinický obraz
- Kůže – projevy jsou variabilní, od jemné a tenké („cigaretový papír“) až po hyperextenzivní („gumovou“) kůži.
- Změny vazů a kloubů – projevy jsou opět variabilní, od mírné hypermobility po nereponovatelné luxace kyčlí a ostatních velkých kloubů. Opakované luxace mohou vést k degenerativním změnám.
- Ostatní – prolaps srdečních chlopní, pes planus, skolióza, u typu VI může dojít k ruptuře bulbu po nejmenším traumatu.


Diagnostika – diagnostika je založena na klinických kritériích a čím dál častěji analýzou DNA. Někdy mohou být modré skléry.
Terapie – kauzální léčba neexistuje. Léčba musí být přísně individuální. Lze zvážit chirurgické napnutí vazů, ale problém je, že v postižených vazech nedrží stehy. U typu IV jsou třeba časté echokardiografické kontroly stavu velkých tepen (přítomnost aneurysmat) a u žen kontroly v těhotenství pro zvýšené riziko ruptury dělohy s krvácením.
Chondrodysplázie (CD)
Incidence je mezi 1 : 2500 – 4000 porodů. Na základě různých rozdílů lze rozeznat > 200 různých typů. Lehké formy se projevují změnami podobnými osteoartróze, většinou je přítomen nanismus s krátkými končetinami, v těžkých případech vysoké čelo s hypoplazií obličeje, rozštěpem patra, změny na očích (katarakta, degenerace sklivce, amoce sítnice). Vlastní dědičnost a genetická postižení jsou složitá (blíže viz Harrison 19e 2511 – 2512). Zvláštním typem je Sticklerův syndrom (vrozená artro-oftalmopatie) s oploštěným obličejem při nevyvinutí kostí obličeje, zrakové a sluchové obtíže. Diagnóza je postavena na klinickém vyšetření, RTG změnách, očním vyšetření a histologickém nálezu. Vzhledem k různorodosti fenotypu je často diagnóza stanovena až specialistou na tuto chorobu. Terapie je symptomatická (náhrada poškozených kloubů, korekce rozštěpu patra, operace katarakty nebo odchlípené sítnice), psychoterapie, pacienti by měli být vedeni k prevenci obezity a vyhnutí se kontaktním sportům.

Marfanův syndrom (MFS)
Epidemiologie – incidence je 1 : 3000 – 5000, dědičnost je v 75 % AD, ve 25 % jde o sporadické mutace. Nejčastěji je mutován gen FBN1 (pro fibrillin – 1).
Klinický obraz
- Postižení skeletu – pacienti bývají vyšší než vrstevníci, s dlouhými, tenkými končetinami i prsty (arachnodaktylií). Dále bývají přítomny deformity hrudníku (pectus excavatum, pectus carinatum) nebo jeho asymetrie, skolióza páteře, ploché nohy a vysoký nárt. Klouby mohou být hypermobilní. Zobrazovací metody mohou odhalit zvětšení neurálního kanálu při ztenčení pediklů a lamin a přední mingokéle (ektázie dury).
- Kardiovaskulární postižení – hlavní příčina morbidity a mortality. Často je přítomen prolaps mitrální chlopně, dilatace aorty a Valsalvova sinu. Rychlost dilatace je nepředvídatelná a může vést k aortální regurgitaci, disekci aorty a její ruptuře (častěji hrudní aorty). Dilatace je urychlena fyzickým a emočním stresem a těhotenstvím.
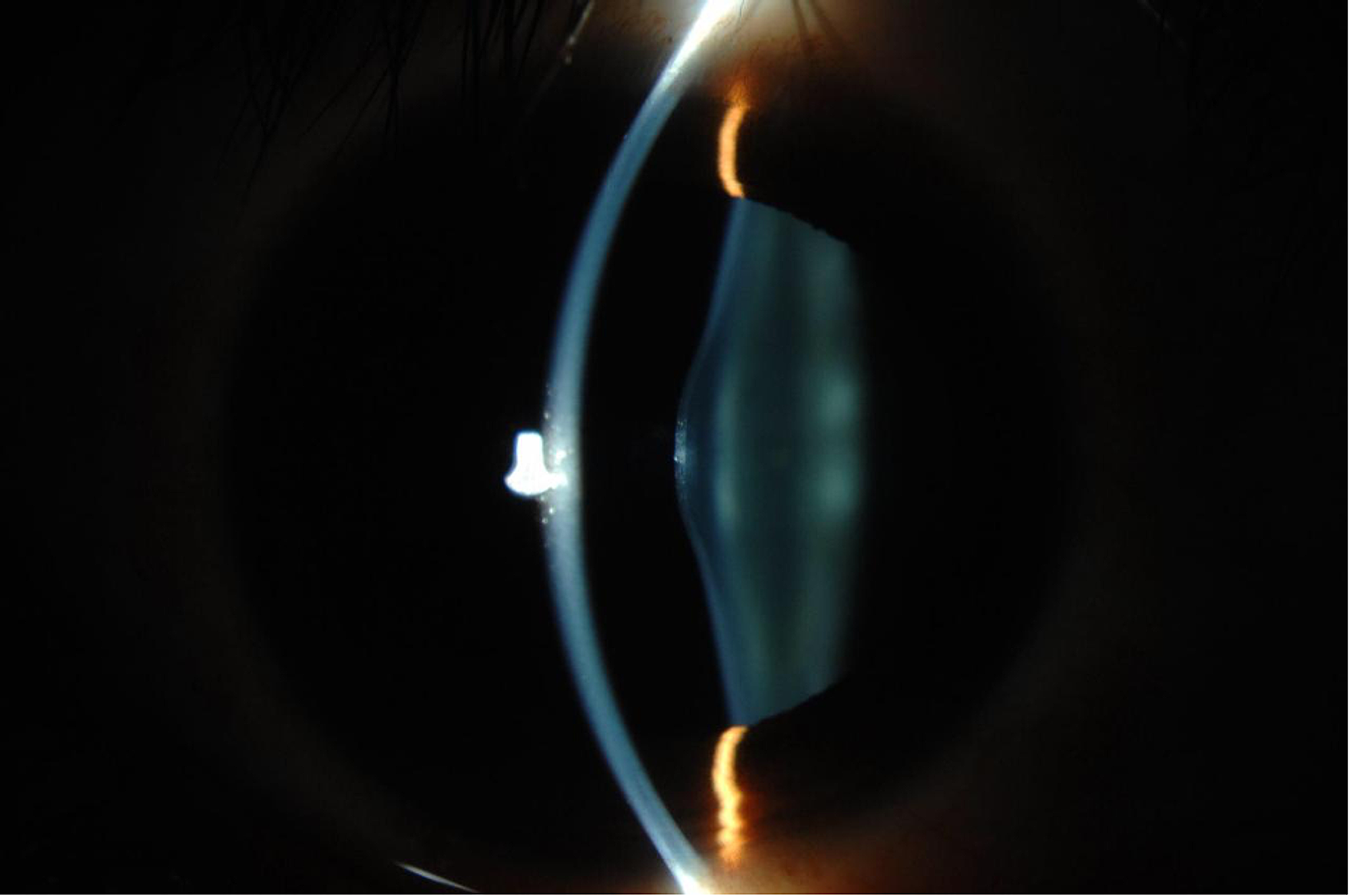
- Oční postižení – s ektopií až luxací čočky.



MFS byl z počátku charakterizován triádou příznaků:
- Dlouhé tenké končetiny s hypermobilitou kloubů (znamení zápěstí, znamení palce)
- Aneurysma aorty.
- Zhoršení vidění díky dislokaci čočky.
K přesné klasifikaci byla vyvinuta Ghentská kritéria https://marfan.org/dx/
Terapie
- ke snížení krevního tlaku a shear stressu a tak i rizika disekce aorty i progrese aneurysmatu indikovány beta blokátory. Při dilataci aorty > 50 mm (u bikuspidní aortální chlopně > 45 mm) je indikována náhrada postižené části aorty
- fyzioterapie a chirurgická korekce skoliózy
- oční operace u vybraných pacientů při dislokaci čočky a amoci sítnice
Epidermolysis bullosa (EB)
Při EB dochází po tření ke vzniku puchýřů jako následek separace jednotlivých vrstev kůže. Incidence choroby je 1 : 50 tisíc. EB je klasifikována podle místa, kde k separaci dochází (toto místo je určeno typem mutace pro určitý protein kůže):
- EB simplex – separace v epidermis, obecně lehčí průběh než následující typy
- Junkční EB – v místě lamina lucida Obr. 8: Epidermolysis bullosa.
- Dystrofická EB – pod sublamina densa (po puchýřích vznikají velké jizvy)
- Kindlerův syndrom – „mix“ separací v různých vrstvách
Diagnóza je dána klinickým obrazem (vznik puchýřů po minimálním traumatu), přesná klasifikace do podtypů na základě imunofluorescence a DNA typizace. Léčba je zatím symptomatická, ve výzkumu je genová a buněčná terapie.

Alportův syndrom (AS)
Incidence je 1 : 10 tisíc porodů, není vždy považován za poruchu pojiva, nicméně bylo prokázáno, že většina pacientů má poruchu kolagenu bazální membrány (typ IV). Jsou rozeznávány čtyři formy:
- klasická – X dědičná (ženy často nediagnostikovány a bývají postiženy méně než muži), tvoří 80 % všech případů AS, trias:
- 1. Hematurie – často progreduje do nefritidy a u některých mužů a ve stáří u žen může způsobit renální selhání.
- 2. Senzoneurální hluchota – zejména v oblasti vysokých tónů (často identifikovatelná pouze na audiogramu a pacient si ji neuvědomuje).
- 3. Lentikonus (konická deformace přední stěny čočky)
- X vázaná – spojená s difuzní leimyomatózou
- AR, AD – obě spojeny s postižením ledvin bez hluchoty i lentikonu