Primární imunodeficity
Úvod – mezi primární imunodeficity (PID) patří více než 350 chorob, které mají většinou Mendelovskou formu dědičnosti. Jsou poměrně vzácné (prevalence 1 : 10 – 20 tisíc) a často poddiagnostikované.
Diagnóza PID – základním znakem PID je přítomnost recidivujících nebo abnormálně závažných infekcí, často se současnou alergií nebo autoimunitou. Vždy je nutná detailní rodinná a osobní anamnéza, při fyzikálním vyšetření je nutné vyšetřit velikost lymfatických orgánů (slezina, uzliny) a hledat specifičtější příznaky jednotlivých poruch:
- časté respirační infekce u poruch protilátkové imunity.
- závažné invazivní bakteriální infekce – porucha protilátkové imunity, defekt komplementu, asplenie.
- virové infekce, recidivující kandidové a oportunní infekce – porucha T lymfocytů.
- kožní infekce a hluboké abscesy – např. u chronické granulomatózní choroby, často u hyper-IgE syndromu.
I. Primární imunodeficity nespecifické vrozené imunity
Jsou vzácné (cca 10 % všech PID).
Těžká vrozená neutropenie
Skupina vrozených chorob, většinou AD dědičných (popsána i AR a X dědičnost), jejichž společným znakem je počet neutrofilů < 0,5 · 109/l krve), a které se většinou manifestují již v dětství. Zatím byly rozpoznány kauzální mutace 16 různých genů (např. ELANE pro elastázu 2, hyperfunkční mutace WASP na chromozómu X vede k Wiskott -Aldrichově syndromu a manifestuje se i monocytopenií). Běžné jsou infekce na povrchu těla a sliznic (rány, respirační trakt), které často diseminují do krve se vznikem sepse a ohrožují život pacienta. Ke stanovení diagnózy je nutné vyšetření kostní dřeně, kde lze prokázat blok granulopoézy nejčastěji na úrovni promyelocytu. Léčba:
- nespecifická zaměřená na důkladnou hygienu, zejména u dětí, důslednou péči o zuby spolu s profylaktickým podáváním trimetoprim-sulfametoxazolu.
- podání G-CSF s dvěma úskalími u nositelů mutace ELANE:
- 1. Někteří jsou k G-CSF rezistentní s nutností alogenní transplantace kostní dřeně,
- 2. u některých v může dojít ke vzniku akutní myeloidní leukémie, která je většinou spojena s hyperfunkční mutací receptoru pro G-CSF.
Asplenie
Primární ageneze sleziny je extrémně vzácná (Ivemarkův syndrom) nebo izolovaně s AD dědičností. Slezina funguje jako fagocytární filtr, který odstraňuje staré a poškozené krevní buňky a cirkulující mikroorganismy. Během průtoku krve sinusoidami sleziny se krevní tok zpomaluje, což napomáhá efektivnímu odstranění defektních erytrocytů a volných i opsonizovaných bakterií makrofágy sleziny. Zvláštní význam má slezina v záchytu a odstranění cirkulujících opouzdřených patogenů, především pak pneumokoků (S. pneumoniae), neboť antigeny polysacharidového pouzdra brání faktorům komplementu v opsonizaci a současně brání i interakci antigen-komplement-makrofág, čímž mikrob uniká fagocytóze. Tyto patogeny jsou efektivně opsonizovány pentamery protilátek třídy IgM, přímo produkovanými paměťovými B-lymfocyty přítomnými ve slezině. Po splenektomii není adekvátně rozvinuta časná fáze specifické vrozené imunitní odpovědi a tito jedinci jsou náchylní zejména k infekcím opouzdřenými mikroorganismy (zejména pneumokok, meningokok a hemofilus). K diagnóze napomáhá přítomnost Howell-Jollyho tělísek v erytrocytech (zbytky erytrocytárního jádra) a je potvrzena sonografií břicha. U dětí do 5 let, dospělých do 1-2 let po splenektomii a s imunosupresí je doporučena antibiotická profylaxe (Penicilin bid) a u všech očkování proti opouzřených mikroorganismům.

Deficit GATA2
Ztrátová mutace genu pro transkripční faktor GATA2 vede k tzv. DCML deficienci (Dendritic Cells, Monocytopenie, Lymfopenie – B a NK). Mimo imunodeficit je zvýšené riziko lymfedému, myelodysplázie a akutní myeloidní leukémie. Infekce jsou život ohrožující a kauzální léčbou je alogenní transplantace kostní dřeně.
LAD
LAD (leukocyte adhesion deficiency) je AR dědičný defekt adheze leukocytu po jeho aktivaci. Tři typy („integrin – rolling – kofaktor integrinu„):
- LAD I – nejčastější forma způsobená mutací genu pro β2 integrin
- LAD II – extrémně vzácný defekt rollingu, který je prvním krokem aktivace
- LAD III – defekt regulačního proteinu kindlinu, který je nutným kofaktorem vazby k β2 integrin. Časté je i krvácení, díky poruše β2 integrinu na trombocytech.
Výsledkem těchto poruch je neschopnost leukocytů adheze a tak i dosažení místa infekce, pacienti jsou tak náchylní k infekcím podobně jako u těžké vrozené neutropenie, navíc je opožděné hojení ran, u novorozenců dlouhá doba do oddělení pahýlu pupečníku. Na diagnózu by mělo být pomyšleno při infekcích bez tvorby hnisu spojených s výraznou leukocytózou (> 30·109/l). Diagnózu lze prokázat imunofluorescencí a funkčními testy. U těžkých forem je indikována alogenní transplantace kostní dřeně.
Chronická granulomatózní nemoc
Chronická granulomatózní nemoc (CGN) je vzácná (prevalence cca 1:200 tisíc) porucha likvidace mikroorganismů neutrofily a makrofágy po jejich fagocytóze většinou díky nedostatku NADPH s následným defektem produkce reaktivních forem kyslíku. V 70 % je X vázaná dědičnost, zbytek je autozomální. Projevuje se hlubokými abscesy lymfatických uzlin, jater a plic, které jsou bohaté na makrofágy a jejichž původci jsou nejčastěji kataláza pozitivní mikroorganismy (např. Staphylococcus aureus a Serratia marcescens), ale často i jiné a dále granulomy, které mohou působit obstrukci (močový měchýř, pylorus) nebo zánět (kolitida). Diagnóza je založena na testech defektní produkce reaktivních kyslíkových radikálů (ROS) neutrofily a makrofágy (průtoková cytometrie). Základem léčby je prevence infekcí (profylaktické podávání trimetoprimu/sulfametoxazolu) a itrakonazolu, při komplikovaném průběhu choroby alogenní transplantace kostní dřeně.
Deficit myeloperoxidázy
AR dědičnost s incideníc 1:2000. Izilovaný deficit myeloperoxidázy je klinicky němý, jelikož ostatní baktericidní mechanismy jej nahradí (mikrobicidní aktivita je sice opožděnám ale je přítomna).
Chédiak – Higashiho syndrom
Vzácná AR choroba s poruchou lyzozomálního transportního proteinu LYST. Neutrofily pacientů mají typicky veliká granula. Typický je nystagmus, albinismus a náchylnost k infekcím, zejména pyogenním (typicky S. aureus).
Vrozená vnímavost k mykobakteriovým infekcím
Tato skupina chorob je charakteristická defektem osy IL-12 a IFN-γ, které jsou zásadní k aktivaci makrofágů, s typickou výraznou vnímavostí k TBC i ostatním mykobakteriím, ale navíc i salmonelovým infekcím. Základem léčby je substituce IFN-γ.
Defekty TLR
Zjištěny u pacientů s četnými virovými a časnými invazivními infekcemi Streptococcus pneumoniae nebo méně často Staphylococcus aureus.
Deficit komplementu
Deficit jednotlivých složek komplementu se může projevit zvýšenou citlivostí k infekcím.
1. Deficit úvodních složek klasické komplementové cesty (C1, C2, C3, C4)
Tendence k závažným bakteriálním pneumoniím nebo meningitidám. Častější výskyt autoimunitních a imunokomplexových chorob (zejména SLE).
2. Deficit terminálních složek komplementové cesty (C5 – C9)
Náchylnost ke komplikovaným bakteriálním infekcím (závažné meningitidy jsou typické zejména pro deficit C9).
3. Deficit složek alternativní komplementové cesty
Deficit faktoru P (properdinu) – X-vázaná dědičnost. Projevuje se závažnými meningokokovými meningitidami, které ale nemají naštěstí tendenci recidivovat.
4. Deficit složek lektinové komplementové cesty
Deficit lektinu vázajícího manózu (MBL) – tnto lektin aktivuje tzv. lektinovou (třetí) komplementovou cestu. Deficit (prevalence 5 – 10 %) je většinou asymptomatický, může se projevovat zvýšenou tendencí k banálním respiračním infekcím, zejména v kojeneckém věku. V dospělosti byla tato porucha častěji prokázána u osob s pneumonií a zejména u pacientů s komplikacemi neutropenie při cytostatické léčbě. Terapie je symptomatická a dále dle závažnosti profylaktické podávání antibiotik.
5. Deficit inhibitorů komplementu
Hereditární angioedém
Epidemiologie – prevalence 1 : 10000 – 50000, věk manifestace nejčastěji okolo puberty.
Etiopatogeneze
1. Primární HAE – choroba je autozomálně dominantně dědičná a je častěji způsobena kvantitativním (HAE I. typu), nebo vzácněji kvalitativním (HAE II. typu) nedostatkem inhibitoru C1. Následkem toho dochází ke spontánní aktivaci klasické komplementové cesty, přičemž její produkty C2b, C3a a C5a mají výrazný vazoaktivní a chemotaktický efekt, dále dochází k aktivaci kininového systému a částečně i hemokoagulace a fibrinolýzy. Největší podíl na vzniku otoků má pravděpodobně hyperprodukce bradykininu. Byl popsán i HAE III. typu, s fenotypem podobným klasickému HAE, ale bez průkazu změnu hladiny nebo funkce C1 inhibitoru. Častěji postihuje ženy a je pravděpodobně také autozomálně dominantně dědičný.
2. Sekundární HAE – provází některé lymfomy nebo systémové choroby pojiva. Klinická manifestace je stejná jako při primární formě. Angioedém bývá i nežádoucím účinkem léčby ACE inhibitory.
Klinický obraz – hlavním projevem choroby je vznik otoku podkoží a podslizničních tkání i po minimálních stimulech (úraz, stomatologický výkon, stres, menstruace). Otok je bledý, nesvědivý, nebolestivý, s pocitem pnutí tkáně (viz obrázek níže). Otok určitých tkání vede ke specifickým komplikacím, u dýchacích cest dušnost s rizikem udušení, u gastrointestinálního traktu koliky, průjmy, zvracení až obraz napodobující NPB, u uretry – retence moči až anurie.

Diagnostika – hlavním cílem diagnózy je průkaz snížené funkce inhibitoru C1. C1 inhibitor je snížený u HAE I. typu nebo normální/zvýšený u HAE II nebo III. typu), C4 je snížený. C1q slouží odlišení primární (normální hladina) a sekundární formy (snížená hladina). Definitivní průkaz primární formy dá genetické vyšetření (mutace genu pro C1 inhibitor).
Terapie
- Režimová opatření a profylaxe – kontraindikace podání ACE inhibitorů, omezení stresu. Při stomatologických výkonech navýšení profylaktické léčby, při instrumentálních výkonech i substituce C1 inhibitoru.
- Farmakoterapie
- Atenuované androgeny (danazol a stanazol) lze použít ke stimulaci syntézy C1 inhibitoru v játrech. Nebezpečím léčby je poškození jater a virilizace pacientek. Inhibitory proteináz (kyselina tranexamová, kyselina ε-aminokapronová) jsou méně účinné.
- Při léčbě akutních otoků se podává koncentrát C1 inhibitoru nebo blokátory receptoru pro bradikinin (ikatibant), v nouzi i čerstvá mražená plazma.
CAVE Antihistaminika a kalcium jsou bez účinku, glukokortikoidy mají minimální účinek.
II. Primární imunodeficity specifické získané imunity
Deficit T lymfocytů
Dělá cca 20 % všech PID. Deficit T lymfocytů má vzhledem k ústřední roli v imunitním systému poměrně zásadní důsledky:
- CD4+ Th1 lymfocyty produkují IFN-γ, který je nutný k aktivaci makrofágů (likvidace intracelulárních patogenů, včetně mykobakterií a salmonel).
- Folikulární CD4+ Th2 lymfocyty v zárodečných centrech jsou zásadní k produkci protilátek, včetně izotypového přesmyku, dále aktivaci eozinofilů (IL-4, IL-5, IL-13) k likvidaci mnohobuněčných parazitů.
- CD4+ Th17 produkují IL-17 a IL-22, které povolávají neutrofily do kůže a plic proti bakteriálním infekcím a mykózám.
- CD8+lymfocyty jsou zásadní k likvidaci virem infikovaných buněk.
- regulační T lymfocyty jsou zásadní k regulaci imunity (komenzálové ve střevě).
SCID
Epidemiologie – SCID (těžký kombinovaný imunodeficit) vede k hlubokému defektu vývoje a tak i absenci T lymfocytů, v 60 % vázaný na X chromozóm (častější u chlapců). Incidence je 1:50 tisíc.
Klinický obraz – SCID se projevuje se ve 3 – 6 měsíci života četnými závažnými infekcemi (recidivujícím soorem, neprospíváním, průjmem, intersticiální pneumonií způsobenou Pneumocystis jiroveci, virózami). Očkování živými infekcemi bývají provázeny až život ohrožujícími komplikacemi. Na diagnózu je nutné pomyslet u pacienta s popsanými obtížemi, často s rodinou anamnézou úmrtí v časném dětství. Může být přítomna šupinatá kožní vyrážka.
Diagnostika – v 90 % je přítomna lymfocytopenie, na RTG S+P chybí stín thymu. K diagnóze je potřeba stanovení počtu T, B a NK buněk. Bližší diagnózu dá imunologické a genetické vyšetření.
CAVE Lymfocytopenie může být úvodem maskována přítomností T lymfocytů pocházejících od matky.
Terapie – základem léčby je alogenní transplantace kostní dřeně.
DiGeorgův syndrom
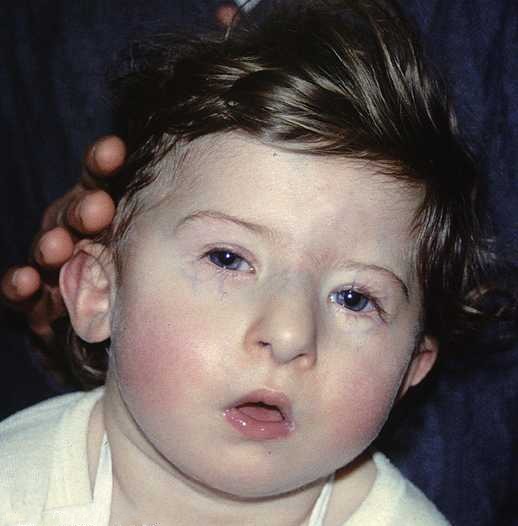
Porucha vývoje 3. a 4. žaberní výchlipky díky deleci 22q11. Mimotechnická pomůcka k zapamatování příznaků: CATCH 22
- Cardiac abnormality (nejčastěji Fallotova tetralogie, vzácněji přerušení aortálního oblouku nebo jen defekt septa síní nebo komor).
- Abnormal face (hypertelorismus, široké philtrum, malá brada, nízce položené a dozadu rotované uši) + mentální retaradace.
- Thymic aplasia (T-lymfocytární imunodeficit různé závažnosti)
- Cleft (rozštěp rtů)
- Hypoparathyreoidismus (v dospělosti se jen vzácně klinicky projevuje, může být zvýšená kazivost zubů)
- 22. chromozóm
Diagnózu potvrdí genetické vyšetření, léčba spočívá v terapii jednotlivých složek syndromu.
Omenn syndrom
Choroba je velice podobná graft vs host reakci v kombinaci se SCID. Typická je nedostatečná heterogenita TCR, která se projevuje unikátním fenotypem (časná erytrodermie + alopecie + hepatosplenomegalie + neprospívání), častá T lymfocytóza, B lymfocytopenie a eozinofilie. Pacienti jsou velice fragilní a vyžadují současnou antibiotickou léčbu i imunosupresi podpořenou nutriční podporou. Léčbou volby je alogenní transplantace kostní dřeně.

Hyper-IgM syndromy
Několik chorob spojených se sníženou hladinu IgG a IgA a normální nebo zvýšenou hladinou IgM . Projevuje se sklonem k respiračním infekcím. Formy:
- X-vázaná – nejčastější, vyvolaná deficitem povrchového ligandu CD40L, mimo respirační infekce jsou i příznaky plynoucí z deficitu T-lymfocytů (pneumocystové pneumonie, kryptosporidiové kolitidy, v době dospívání častá primární sklerotizující cholangoitida)
- Recesivní – způsobena deficitem enzymu AID (activation induced deaminase), poruchou exprese CD40 apod.
Wiskott – Aldrichův syndrom
Choroba s X-vázanou dědičností s incidencí 1:200 tisíc, způsobená mutací WASP genu. která se projevuje klinickým trias TIA:
Trombocytopenie s krvácením + Imunodeficit + Atopický ekzém
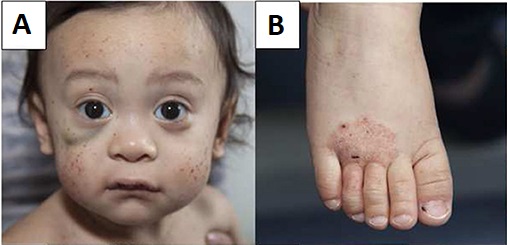
Pokud se nemocní dožijí dospělosti, jsou ohroženi lymfomy a Ig-A nefropatií.
Terapie záleží na závažnosti obtíží, splenektomie zmírní trombocytopenii, ale je zatížena výrazným rizikem infekcí, dobrý efekt má alogenní transplantace kostní dřeně.
Důležitou skupinou chorob jsou nemoci spojené s instabilitou chromozómů (ataxia telangiectasia, Nijmegenský syndrom,Bloomův syndrom).
Hyper IgE syndrom (Jobův syndrom)
Způsoben mutací genu pro intracelulární signalizační protein STAT3 vede k poruše vývoje Th17 lymfocytů. Typické příznaky:
- Ekzém
- Výskyt hlubokých abscesů v podkoží a plicích, někdy s minimální zánětlivou odpovědí („studený absces“). Nejčastějším vyvolávajícím agens je S. aureus a kandidy.
- Nemocní mají typicky hrubé rysy obličeje.
- Zvýšená lomivost kostí, častější skolióza, problémy s chrupem.
- Vysoká hladina IgE
- Eozinofilie

Typický klinický obraz + vysoká hladina IgE + genetické vyšetření na mutaci STAT3. Obtížné je odlišení atopické dermatitidy (ekzém + zvýšené IgE), na rozdíl od ní přítomny hluboké abscesy, typická facies a zubní a kostní změny. Léčba je symptomatická léčba a dále profylaxe infekcí (protistafyloková antibiotika).
Typický vzhled pacientů zde: https://healthjade.net/job-syndrome/
Deficit B lymfocytů
Postižení B lymfocytů patří mezi nejčastější PID (cca 60 – 70 %). Je zde řada míst výskytu:
- pentamery IgM jsou přítomny intravaskulárně a na povrchu sliznic.
- IgA jsou secernovány zejména MALT
- IgG může difundovat volně extravaskulárně
Postižení specifické humorální imunity je nejčastěji spojené s recidivujícími pyogenními bakteriálními infekcemi (pneumonie, sinusitidy). Chybění protilátek (agamaglobulinémie) bývá doprovázena invazivními enterovirovými infekcemi. Tyto PID se projevují jen zřídka před 6 měsíci věku (do té doby dočasně chrání mateřské IgG protilátky předané transplacentárně). Diagnóza je dána nízkou hladinou imunoglobinů, bližší informaci podá typizace imunoglobulinů, vyšetření kostní dřeně a fenotypizace B lymfocytů.
X-vázaná hypogamaglobulinémie (Brutonova hypogamaglobulinémie)
Prevalence choroby je 1: 50 – 100 tisíc. Jde o X recesivně dědičnou mutaci genu pro Btk (Brutonova tyrozin kináza), enzymu nutného pro vývoj B-lymfocytů. U postižených chlapců proto nejsou v krvi přítomné zralé B-lymfocyty. Manifestace začíná v časném dětství komplikovanými respiračními infekcemi, časté i průjmovité a neurologické infekce. Oproti CVID nejsou přítomny autoimunitní choroby ani granulomy. Léčbou volby je substituce imunoglobulinů a adekvátní léčba infekcí. Po zavedení substituce vedou pacienti plnohodnotný život a dožívají se dospělosti.
CVID
Definice – běžný variabilní imunodeficit je podle ESID (Evropská společnost pro imunodeficity) definován jako snížená hladina sérových imunoglobulinů v kombinaci s porušenou specifickou protilátkovou odpovědí a to zejména po specifickém antigenním stimulu (typicky očkování).
Epidemiologie – prevalence je 1:20 tisíc, choroba se může manifestovat kdykoliv, nejčastěji ve věku 10 – 30 let.
Etiologie – heterogenní skupina chorob genetické etiologie (pravděpodobně celá řada různých mutací). U části nemocných lze zaznamenat podobně postižené příbuzné. V anamnéze může být selektivní deficit IgA, který do plně vyjádřeného CVID může progredovat (častá mutace genu TNFRSF13B, kódujícího membránový aktivační receptor B lymfocytů TACI, asi nejde o kauzální příčinu).
Patogeneze – přítomna řada abnormalit funkce i počtu T- i B-lymfocytů a někdy i APC, přičemž zásadní je blokáda produkce protilátek B-buněk. Ne všechny příznaky jsou vyvolány poruchou produkce protilátek, důležitá je i porucha regulační funkce T-lymfocytů, která se může podílet na častém vzniku autoimunitních chorob.
Klinický obraz
- 1. Respirační infekce – u některých nemocných je anamnéza častých infekcí v dětství, u jiných dospělých je vznik infekcí de novo. Ve 2. nebo 3. dekádě dochází ke vzniku opakovaných a prolongovaných bronchitid, otitid, sinusitid a bronchopneumonií s postupným vznikem bronchiektázií nebo plicní fibrózy. K virovým infekcím nejsou pacienti zvýšeně náchylnější (mimo enteroviry). Nejčastějšími etiologickými agens jsou opouzdřené mikroorganismy (zejména Haemophilus influenzae, Streptococcus pneumoniae, méně často i Staphylococcus aureus, Moraxella catarrhalis). Kolonizace Pseudomonas aeruginosa je obávanou komplikací pacientů dlouhodobě léčených kortikoidy.
- 2. Gastrointestinální příznaky – od brzkého věku přítomna atrofická gastritida s perniciózní anémií, běžné i nespecifické střevní záněty. Nejčastější příčinou chronických průjmů je infekce Giardia lamblia, méně často kampylobakterové, rotavirové a salmonelové infekce. Často se neprokáže žádné etiologické agens (někdy se hovoří o CVID kolitidě).
- 3. Muskuloskeletální příznaky – častěji přítomny systémové choroby pojiva a artritidy (infekční i neinfekční).
- 4. Ostatní – častěji přítomny:
- autoimunitní choroby – atrofická gastritida s perniciózní anémií, idiopatické střevní záněty, ITP, hemolytické anémie, autoimunitní neutropenie a další
- malignity (zejména lymfomy a karcinom žaludku)
- lymfadenopatie, u 30 % pacient splenomegalie
- nekaseifikující granulomy připomínají sarkoidózu neznámé etiologie, někdy solitární, častěji ale mnohočetné (plíce, játra, slezina apod.)
- enterovirové infekce – nejčastěji meningoencefalitida (chronický průběh trvající měsíce až roky, obvykle s fatálním koncem s příznaky emoční lability, změny osobnosti, poruchy kognitivních funkcí, někdy akutní forma s bolestmi hlavy a křečemi)
Diagnostika – stanovení na základě typické anamnézy a laboratorních nálezech:
- 1. Anamnéza – komplikované respirační infekce (často u člověka, který jimi dříve netrpěl)
- 2. Laboratorní nález
- snížené IgG a IgA, IgM
- porucha specifické protilátkové odpovědi po antigenním stimulu (nejčastěji očkování vakcínou proti tetanu nebo pneumokokovi)
- vyšetření lymfocytů je normální (na rozdíl od Brutonovy hypogamaglobulinémie)
CAVE Zdaleka ne každé snížení hladiny imunoglobulinů = CVID.
Terapie
- Základem terapie je substituce imunoglobulinů, zejména IgG (poločas 21 dnů, IgA a IgM mají poločas příliš krátký).
- Agresivní léčba bakteriálních infekcí, původci bývají zejména opouzdřené bakterie (H. influenzae, Str. pneumoniae, S. aureus), čemuž je třeba upravit léčbu. U některých pacientů je vhodná sezónní antibiotická profylaxe (lék volby cotrimoxazol, někdy indikováno střídání jednotlivých preparátů). Při dlouhodobé profylaxi je nutné občasná kontrola kvasinek ve sputu s event. přidáním antimykotika. Při chronických průjmech je třeba vyloučit, popř. léčit lambliózu (metronidazol).
- Léčba granulomů kortikosteroidy.
Komplikace – u velké části pacientů se průběh komplikuje vznikem bronchiektázií nebo plicní fibrózy. Nezvladatelné průjmy mohou někdy vést až k malnutrici.
Selektivní deficit IgA
Prevalence v ČR je 1:400, etiologie není známa (časté nahromadění deficitu v jedné rodině potvrzuje genetický podklad). U některých osob lze zaznamenat výrazné kolísání hladin IgA (od téměř neměřitelných po téměř normální). Choroba je geneticky podobná CVID (často do ní i přechází). Existují i farmakologicky indukované formy (fenytoin,cyklosporin, sulfasalazin apod.). Většina postižených je asymptomatická, u některých jsou příznaky slabšího imunodeficitu (častější banální infekce), který se objevuje v předškolním věku a poté postupně mizí. Jde tedy zejména o laboratorní odchylku. Je predispozice ke vzniku autoimunitních chorob (zejména celiakie, dále juvenilní revmatoidní artritidy a systémového lupus erythematodes). Vzácněji bývá tento imunodeficit celoživotní, u takových pacientů lze prokázat i deficit některé podtřídy IgG (nejčastěji IgG2). Diagnostika je dána opakovaným změřením nízké hladiny IgA při normální hladině ostatních protilátek. Kauzální léčba není známá, při častých infekcích lze užít bakteriální imunomodulátory.
CAVE V případě celiakie je screeningové vyšetření IgA proti tkáňové transglutamináze a endomyziu hladkého svalstva negativní (falešně negativní test).
U některých osob s absolutním deficitem IgA je přítomno chybění imunologické tolerance IgA. Po podání některých přípravků (např. erymasy, mražené plazmy, imunoglobulinů) a vzácně během těhotenství se do organismu dostane malé množství IgA s následným rozvojem anafylaktické reakce. Tyto reakce bývají poměrně vzácné, ale u pacientů s IgA deficitem je s tímto rizikem třeba počítat.
Deficit podtříd IgG
IgG v plazmě je ve 4 podtřídách, někdy dochází ke snížení produkce jedné ze tříd s tendencí ke vzniku infekcí způsobených opouzdřenými bakteriemi. Stav bývá někdy závažný s nutností substituce imunoglobulinů.
III. Sekundární imunodeficity
Imunodeficity, které vznikají důsledkem jiných chorob, jejich léčby, popřípadě jiných definovaných stavů. Jejich četnost je ve srovnání s primárními imunodeficity nesrovnatelně vyšší, projevy ovšem bývají zároveň mnohem mírnější (mimo AIDS a sekundární agranulocytózu). Patogeneze bývá většinou multifaktoriální a komplexní, proto jsou rozdělovány podle vyvolávající příčiny.
1. Sekundární hypogamaglobulinémie
Poměrně dobře definovaná skupina chorobných stavů s typickou symptomatologií humorálního imunodeficitu (opakované a komplikované respirační infekce). Dle příčiny:
- 1. Snížená tvorba imunoglobulinů
- hematologické malignity (CLL, B-lymfomy, mnohočetný myelom)
- polékově (sulfasalazin, koloidní zlato, hydantoin)
- 2. Ztráty imunoglobulinů
- močí (nejčastěji nefrotický syndrom), dochází k poklesu hladiny IgG
- stolicí (exsudativní enteropatie, hlavně Ménétriérova choroba a střevní lymfangiektázie, méně nespecifické střevní záněty), klesají všechny třídy imunoglobulinů
Snížená tvorba imunoglobulinů se může projevovat zvýšenou náchylností k respiračním infekcím, jejich ztráty bývají obvykle klinicky němé. Léčebně lze použít substituci imunoglobuliny, potřebné dávky bývají nižší než při primárních hypogamaglobulinémiích. Důležitá je včasná antibiotická léčba infekcí.
2. Imunodeficit po splenektomii
Ve slezině se tvoří protilátky, dále zde lienální makrofágy fagocytují cirkulující opsonizované částice. Obě funkce jsou zástupné, proto pacienti většinou netrpí zvýšenou náchylností k banálním infekcím. Hlavní rizikem je OPSI (overhelming postsplenectomy infections).
OPSI – nemocní po splenektomii jsou ohrožení prudce se rozvíjející sepsí s vysokou mortalitou. Nejčastějším vyvolávajícím agens je Streptococcus pneumoniae, v menší míře se uplatňují Neisseria meningitidis, Haemophilus influenzae, stafylokoky, Escherichia coli. Každého pacienta po splenektomii (nejlépe ještě před výkonem v případě plánované splenektomie) je důležité očkovat proti pneumokokům, optimálně ještě proti Neisseria meningitidis a Haemophilus influenzae. U dětí do 5 let, dospělých do 1-2 let po splenektomii a s imunosupresí je doporučena antibiotická profylaxe (Penicilin bid).
3. Imunodeficit při metabolických chorobách
Velmi variabilní, přesto jsou některé imunologické komplikace relativně typické:
- Urémie – komplexní porucha nespecifické i specifické imunity se zvýšená náchylnost k stafylokokovým, mykobakteriálním a HBV infekci (snížená schopnost eliminace viru často vedoucí k chronickému nosičství).
- Jaterní selhání – porucha proteosyntézy včetně složek komplementu i dalších opsoninů. Přítomny kvantitativní (hypersplenismus) i kvalitativní změny granulocytů:
- hypergamaglobulinémie vznikající zvýšeným průnikem antigenů a ostatních stimulantů syntézy imunoglobulinů střevní stěnou do krevního oběhu a jejich nedostatečnou eliminací játry
- predispozice ke vzniku flegmón a pyodermií
- Diabetes mellitus – postihuje funkci T lymfocytů i granulocytů (baktericidní mechanismy a chemotaxe). Změny jsou nejvýraznější u špatně léčeného event. neléčeného diabetu. Zvýšená náchylnost ke vzniku pyodermií se zhoršením hojení ran a tendencí k rozvoji gangrén a ulcerací a kožních plísňových onemocnění.
4. Imunodeficit při malnutrici
V našem regionu dochází k malnutrici zejména při poruše příjmu potravy (mentální anorexie, různé formy diet), onemocnění GIT nebo alkoholismu. Naopak, z celosvětového hlediska je nejvýznamnější příčinou malnutrice podvýživa.
- Deficit vitamínu a minerálů – z hlediska imunodeficitů se negativně projevuje deplece vitamínů skupiny B, C, D, železo, měď, zinek, selen, mangan. Klinické syndromy vyplývající z jejich deficitu (např. skorbut při nedostatku vitamínu C nebo acrodermatitis enteropathica) jsou v našich končinách raritní.
- Kwashiorkor – lze sice prokázat normální nebo mírně zvýšené hladiny imunoglobulinů (způsobeny intenzivní stimulací imunitního systému při chronických nebo recidivujících infekcích), specifická protilátková i T-buněčná reakce je ale oslabena.
- Mentální anorexie – postižena zejména T-buněčná reakce. Přestože pacientky netrpí zvýšenou incidencí banálních infekcí, může se u nich projevit fatálně probíhající sepse a pneumonie.
5. Imunodeficit při virových onemocněních
Řada virů má schopnost tlumit imunitní systém. Nejvýznamnější představitelé:
- HIV (snížení počtu pomocných CD4+ lymfocytů), méně CMV, EBV a některé další herpetické viry (interference s funkcí APC a cytokinů)
- spalničky (pokles počtu T- i B-lymfocytů, porucha funkce monocytů-makrofágů s rizikem smrtelné oportunní infekce)
- všeobecně všechny virózy vedou k lokálnímu oslabení slizničních bariér i zhoršení specifické imunity s tendencí ke vzniku významných bakteriálních superinfekcí, zejména závažných pneumonií
6. Imunodeficit při stresu
Aktivace hypofýzo-adrenální osy ovlivňuje negativně celou řadu imunitních reakcí. Glukokortikoidy alterují zejména funkci T- a B-lymfocytu, ovlivněny jsou i NK-buňky. Dlouhodobý stres ovlivňuje vnímavost k infekcím a snad i zvyšuje tendenci k nádorovému bujení.
7. Imunitní systém ve stáří
U osob vysokého věku jsou přítomny některé abnormality imunitního systému:
- ↑ IgG a IgA
- ↓ IgM
- množství T-lymfocytů nebývá výrazně změněno, dochází ale k poruše jejich funkce
- je zachována dobrá sekundární odpověď, primární imunitní odpověď je zhoršená (jinými slovy, senioři umí dobře reagovat na infekce, se kterými se dříve setkali, s novými antigeny si již tak dobře poradit nedokážou).
Nebezpečné jsou virové infekce s možnou bakteriální superinfekcí, podobně je zvýšené riziko exacerbací ostatních chorob. Preventivně je doporučována vakcinace proti pneumokokům, chřipce a dodržovat očkovací schémata proti tetanu.
Zdroje
Doporučený postup péče o pacienty s porušenou nebo zaniklou funkcí sleziny (hyposplenismem/asplenií), dostupné online na: https://www.infekce.cz/DopOPSI13.htm
Harrison’s Principles of Internal Medicine, 20e, New York, NY: McGraw-Hill; 2019