Metabolismus bilirubinu – bilirubin je koncový produkt degradace hemu (70 – 90 % vzniká degradací hemoglobinu ze stárnoucích erytrocytů). Bilirubin vznikající na periferii je plazmou transportován (díky nerozpustnosti ve vodě vázaný na albumin) do jater, kde je transportován napříč hepatocytem, což se děje čtyřmi odlišnými, ale navzájem provázanými kroky:
- 1. Vychytávání bilirubinu hepatocyty je zprostředkováno transportéry, které se ale zatím nepodařilo identifikovat.
- 2. Intracelulární vazba – v hepatocytu je nekonjugovaný bilirubin vázán ke glutathion-S- transferáze.
- 3. Konjugace – bilirubin je konjugován s jednou nebo dvěma částmi kyseliny glukuronové pomocí UDP-glukuronyl transferázy za vzniku bilirubin mono- nebo diglukuronidu, který se stává vysoce rozpustným ve vodě (konjugace je nutná k transportu přes membránu žlučového pólu hepatocytu do žlučových kapilár).
- 4. Vylučování – mono- a diglukuronidy bilirubinu jsou vylučovány přes kanalikulární membránu žlučového pólu hepatocytu do žlučových kapilár pomocí ATP dependentního transportéru MRP2 (multidrug resistence – associated protein 2). Jeho mutací vzniká Dubin – Johnsonův syndrom.
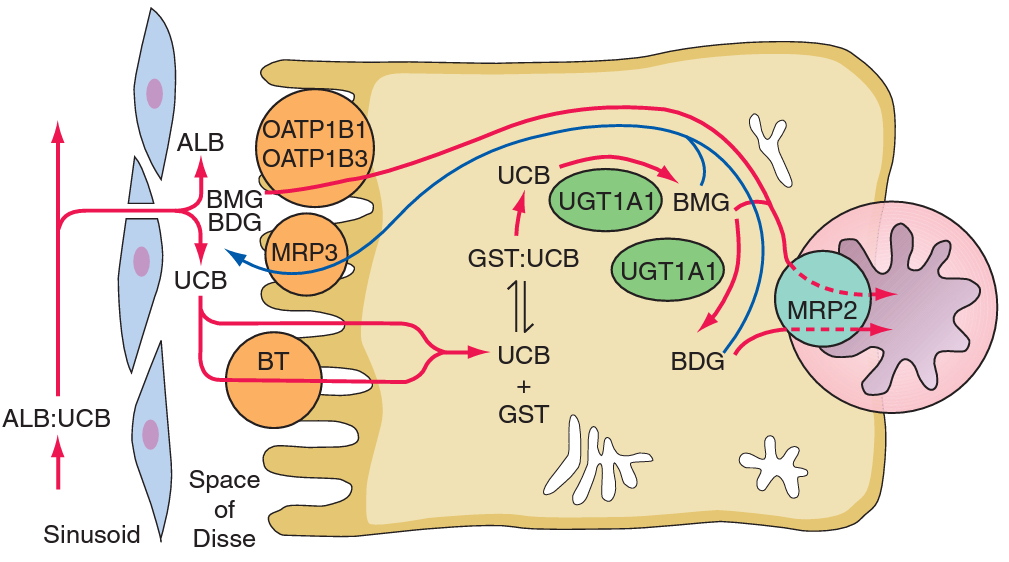
Po exkreci do žluče je konjugovaný bilirubin vylučován do duodena a tudy pokračuje aborálně gastrointestinálním traktem bez reabsorpce. Značný podíl je konvertován bakteriálním metabolismem na solubilní, bezbarvý urobilinogen, který podstupuje enterohepatální cyklus. Není vychytáván játry a přestupuje přímo do systémové cirkulace, odkud je odstraňován ledvinami. Nekonjugovaný bilirubin většinou nedosahuje střeva (mimo novorozeneckého ikteru nebo těžké nekonjugované hyperbilirubinémie – Crigler – Najjarův syndrom typ I).
Nekonjugovaný bilirubin nemůže být vyloučen močí, je v plazmě pevně vázán na albumin. Konjugovaný bilirubin je ledvinami filtrován a vylučován močí.
Stavy vedoucí k nekonjugované hyperbilirubinémii
1. Zvýšená produkce bilirubinu
Hemolýza – zvýšená destrukce erytrocytů vede ke zvýšení množství bilirubinu a nekonjugované hyperbilirubinémii, která bývá většinou mírná. Kostní dřeň je schopna maximálně osminásobného zvýšení produkce erytrocytů jako kompenzace hemolýzy. Z toho vyplývá, že hemolýza sama o sobě nemůže dát vzniku hodnotám bilirubinu > 68 μmol/l (vyšší hodnoty znamenají současnou jaterní dysfunkci). V případě čisté hemolýzy je hodnota konjugované frakce < 15 % a jde o nekonjugovanou hyperbilirubinémii. V případě přítomné systémové choroby, která vede k jaterní dysfunkci se může na celkové hyperbilirubinémii podílet i konjugovaná frakce. Dlouhodobá hemolýza vede k precipitaci bilirubinových solí v žlučovém stromě i žlučníku, což vede k pigmentové cholelitiáze.
Inefektivní erytropoéza – v průběhu zrání erytrocytů se může uvolňovat malé množství hemoglobinu jednak při procesu denukleace, jednak je část erytrocytů likvidována přímo v kostní dřeni. Tyto procesy jsou normálně provázeny pouze malou produkcí bilirubinu. V průběhu různých chorob (thalassemia major, megaloblastová anémie, kongenitální erytropoetická porfyrie, otrava olovem, dyserytropoetické anémie) roste množství bilirubinu, který vzniká v průběhu inefektivní erytropoézy až na úroveň 70 % celkového bilirubinu, což může vést k lehkým formám nekonjugované hyperbilirubinémie.
Ostatní – degradace hemoglobinu z extravaskulárních kolekcí erytrocytů (masivní infarkty, hematomy) mohou vést k přechodné nekonjugované hyperbilirubinémi.
2. Snížená jaterní clearance bilirubinu – Gilbertův syndrom, léky (rifampicin a řada cholecystografických kontrastních látek).
3. Porucha konjugace
Fyziologický ikterus novorozenců – bilirubin produkovaný plodem je odstraňován placentou a eliminován játry matky. Bezprostředně po narození musí novorozenecká játra převzít zodpovědnost za filtraci a exkreci bilirubinu, ale aktivita UGT1A1 je nízká a alternativní cesty umožňují průchod nekonjugovaného bilirubinu do střeva. Protože je střevní flóra, která konvertuje bilirubin na urobilinogen také nedostatečně vyvinutá, následuje enterohepatická cirkulace nekonjugovaného bilirubinu. Díky tomu většina novorozenců tak trpí nekonjugovanou hyperbilirubinémie 2. – 5. den po porodu s peakovou hladinou většinou mezi 85 – 170 μmol/l a klesá k normálním hodnotám do 2 týdnů, jak postupně dozrávají mechanismy degradace. Nezralost novorozenců bývá spojena i s nezralostí jaterních funkcí což může vyústit do výraznější nekonjugované hyperbilirubinémie. Rychlý nárůst nekonjugovaného bilirubinu nad 340 μmol/l ohrožuje novorozence bilirubinovou encefalopatií neboli jádrovým ikterem. Za těchto okolností, bilirubin prochází nezralou hematoencefalickou bariérou a precipituje zejména v bazálních gangliích s následným výrazným neurologickým deficitem až smrtí. Léčbou jsou fototerapie (převádí bilirubin do rozpustných fotoizomerů, které jsou vylučovány do žluče) a výměnné transfúze.
3a Získané poruchy konjugace
- pokročilá hepatitida nebo cirhóza vede k mírné redukci konjugační kapacity, postižení této části metabolismu je menší než např. vylučování bilirubinu na žlučovém pólu hepatocytu.
- léky – např. pregnandiol, chloramfenikol, gentamycin – mohou způsobit nekonjugovanou hyperbilirubinémii inhibicí aktivity UGT1A1.
- inhibice UGT1A1 novorozence
- některé mastné kyseliny přítomné v mateřském mléce, mohou inhibovat aktivitu UGT1A1 novorozence se vznikem nekonjugované hyperbilirubinémie. U těchto novorozenců může hrát roli zrychlená enterohepatální cirkulace.
- Lucey – Driscollův syndrom – vzácná přechodná familiární novorozenecká hyperbilirubinémie, kdy se z matky do novorozence dostává inhibitor UGT1A1.
3b Vrozené poruchy konjugace
Crigler – Najjarův syndrom, typ I – poprvé popsán v roce 1952, jeho prevalence je 1:1-1,5 miliónu. Mnozí pacienti žijí ve společensky nebo geograficky izolovaných komunitách, kde jsou běžné příbuzenské svazky. AR dědičná mutace genu UGT1A1 se vznikem defektu v konjugaci bilirubinu kyselinou glukuronovou (kromě bilirubinu chybí konjugace i řady léků). Před vynálezem fototerapie zemřela většina pacientů s CN-I na bilirubinovou encefalopatii (kernikterus) v ranném dětství. Projevuje se nekonjugovanou hyperbilirubinémií (340 – 765 μmol/l), která se objevuje již v novorozeneckém věku. Ostatní jaterní testy (ALT, AST, ALP) jsou normální a není přítomna hemolýza. V moči se nenachází žádný bilirubin, ve žluči chybí konjugovaný bilirubin. Jaterní histologie je normální (mimo občasný záchyt žlučových zátek v žlučových kanálcích). Není přítomna aktivita genu UGT1A1 v jaterní tkání (nereaguje na podání fenobarbitalu nebo jiných induktorů enzymů). Nekonjugovaný bilirubin se hromadí v plazmě, odkud se odstraňuje velmi pomalu alternativními cestami do žluče a do tenkého střeva. Základem léčby je transplantace jater.
Crigler – Najjarův syndrom, typ II – choroba byla uznána jako samostatná jednotka v roce 1962. Množství mutací vysvětluje různé spektrum závažnosti choroby. Na rozdíl od CN-I je průměrná koncentrace bilirubinu nižší, kernikterus je přítomen jen zřídka, je přítomna snížená aktivita UGT1A1 (typicky 10 %) a bývá přítomna reakce na fenobarbital (jeho podání vede k poklesu bilirubinu o > 25 %), který je doporučen jako léčba.
Gilbertův syndrom – prevalence choroby je 8 %, muži jsou 1,5 – 7x častěji postiženy než ženy, záchyt je nejvíce v pubertě nebo dospělosti. Přítomna je AD dědičnost s variabilní expresivitou a typické je snížení aktivity UGT1A1 na 10 – 35 % normy se snížením clearance bilirubinu na cca 1/3 normy. Projevuje se lehkou nekonjugovanou hyperbilirubinémií bez jiných známek choroby. K progrese hyperbilirubinémie vede stres, únava, užití alkoholu, malnutrice a infekce, zatímco zvýšení kalorického příjmu nebo aplikace enzymového induktoru (rifampicin) má za následek snížení hladiny bilirubinu až normalizaci hodnot. Laboratorně je celkový bilirubin většinou ˂ 51 μmol/l (většinou nedosahuje hladin jako u CN-II) a podáním fenobarbitalu se dosáhne normalizace hodnot (selhání upozorní na přítomnost jiné vady). Ostatní jaterní testy (enzymy, albumin, protrombinový čas, jaterní biopsie – u některých pacientů přítomna lehká depozita lipofuscinu) jsou normální. Žluč obsahuje charakteristicky zvýšenou frakci monoglukuronidu bilirubinu. Léčba není potřeba. Metabolismus většiny léků, které jsou metabolizovány pomocí glukuronidace se zdá být normální. Výjimky:
- cytostatikum irinotekan (CPT-11), jehož aktivní metabolit (SN-38) je glukuronizován specificky bilirubin-UDP glukuronosyltransferázou. Podávání irinotekanu u pacientů s GS vedlo k několika intoxikacím (průjem, myelosuprese).
- dle několika studií je i abnormální metabolismus mentolu, estradiol benzoátu, paracetamolu, tolbutamidu a rifampicinu. Přestože výsledky těchto studií jsou sporné a nebyly zaznamenány žádné klinické komplikace, je třeba zvýšené opatrnosti u pacientů s GS, pokud jim předepisujeme léky, které jsou metabolizovány pomocí glukuronidace.
- inhibitory HIV proteázy indinavir a atazanavir přímo inhibují UGT1A a dále zhoršují jeho funkci u pacientů s GS.
Stavy vedoucí ke smíšené nebo konjugované hyperbilirubinémie
Smíšená hyperbilirubinémie vzniká při přidružených jaterních chorobách (např. akutní hepatitida, choledocholitiáza). Při extra- i intrahepatální cholestáze vzniká konjugovaná hyperbilirubinémie a nelze na základě výše nebo poměru konjugovaného a nekonjugovaného bilirubinu určit zda je příčina obstrukce v játrech nebo mimo játra.
Hlavním důvodem stanovení celkového a konjugovaného bilirubinu je primární odlišení hepatocelulárních a obstrukčních poruch (smíšená nebo konjugovaná hyperbilirubinémie) od vrozených a hemolytických poruch (nekonjugovaná hyperbilirubinémie).
1. Vrozené defekty jaterní exkrece
Dubin-Johnsonův syndrom – AR dědičná porucha funkce MRP2. Benigní, relativně vzácná porucha, typicky s mírnou, zejména konjugovanou, hyperbilirubinémií. Pacienti s DJS jsou většinou asymptomatičtí, přestože někteří mohou mít lehké, nespecifické obtíže. Hladina bilirubinu může výrazně stoupnout při interkurentních infekcích, užití hormonální antikoncepce nebo těhotenství. Vzhledem ke konjugované hyperbilirubinémii je přítomná bilirubinurie, jiné příznaky nejsou většinou přítomny (někteří pacienti mohou mít hepatosplenomegalii). Základním rysem je akumulace tmavého, zrnitého materiálu v lyzozomech centrilobulárních hepatocytů, což dává játrům černou barvu. Celkový bilirubin je většinou 34 – 85 μmol/l (během zhoršujících faktorů může stoupnout i na 340 – 430 μmol/l a bývá variabilní i interindividuálně). Ostatní laboratorní testy jsou v normě. Terapie není potřeba.
Typicky přítomny abnormality ve vylučování močového koproporfyrinu (za normálních okolností je v moči přítomno 75 % koproporfyrinového izomeru III). U pacientů s DJS je celkový obsah koproporfyrinů normální, ale ˃ 80 % je izomer I. Heterozygoti vykazují tuto abnormalitu pouze částečně. Molekulová podstata tohoto jevu zůstává neznámá.
Rotorův syndrom – benigní, AR dědičné onemocnění je klinicky podobné DJS, je ale ještě vzácnější. Jedinou biochemickou abnormalitou je elevace celkového bilirubinu, zejména jeho konjugované frakce, což je spojeno s bilirubinurií. Na rozdíl od DJS jsou játra pacientů s Rotorovým syndromem bez pigmentací a jeví se zcela normální.
Benigní rekurentní intrahepatální cholestáza (BRIC) – vzácná AR dědičná choroba charakterizována rekurentními atakami pruritu a ikteru. Typická epizoda cholestázy začíná mírnou nevolností a elevací ALT a AST, následuje rapidní nárůst ALP a konjugovaného bilirubinu spolu s ikterem a pruritem. Prvních několik epizod může být špatně diagnostikováno jako akutní virová hepatitida. Mohou trvat týdny a měsíce a jsou střídány kompletními klinickými i biologickými remisemi, které mohou trvat měsíce až roky. Choroba nevede ani k cirhóze ani k end stage poškození jater. Klasifikace:
- BRIC1 – porucha genu FIC1, který je exprimován zejména v tenkém střevě a jen málo v játrech. Protein FIC1 je pravděpodobně členem rody P-typu rodiny ATPáz, které transportují aminofosfolipidy přes buněčné membrány různých buněk. Jeho vztah k patofyziologii poruchy zůstává neznámý.
- BRIC2 – mutace BSEP (bile salt excretory protein). Podobně i zde zůstává jeho vztah k patofyziologii poruchy neznámý.
Progresivní familiární intrahepatální cholestáza (FIC) – tři fenotypově podobné, syndromy:
- FIC 1 (Bylerova choroba) se projevuje v dětství cholestázou, která může být zpočátku epizodická. Na rozdíl od BRIC progreduje do malnutrice, růstové retardace a end-stage jaterního selhání během dětství. Podkladem je mutace FIC1, jejíž patofyziologický vztah ale zůstává neznámý.
- FIC 2 je progresivní, spojená s mutací bile salt excretory proteinu, který je hlavním transportérem žlučových kyselin pro žlučové kyseliny na žlučovém pólu hepatocytu. Některé mutace tohoto proteinu mohou být spojeny s BRIC2.
- FIC 3 je progresivní, spojená s mutací MDR3, proteinu, který je nutný pro normální hepatocelulární exkreci fosfolipidů na žlučovém pólu hepatocytu.
Přestože všechny 3 typy mají podobný fenotyp, pouze FIC3 je spojen s elevací GGT. U BRIC a FIC 1 a 2 je hladina GGT normální nebo jen lehce zvýšená.