Definice – skupina poruch vznikající agregací a depozicí bílkovinných substancí ve formě nerozpustných amyloidových fibril v antiparalelním β-uspořádání, uložených extracelulárně v podobě homogenních amorfních hmot, tj. amyloidu (termín poprvé použil Rudolf Virchow v roce 1854, protože mu tato depozita pod mikroskopem připomínala škrob, latinsky amylum). Hromadění amyloidu vede k postižení funkce jednotlivých orgánů.
Klasifikace – založena na biochemickém původu mateřského proteinu. Standardní nomenklatura je označována jako AX (A = amyloid, X = amyloidový protein):
- AL – volné lehké řetězce (může být asociována s lymfomem nebo myelomem).
- AA – sérový amyloid A (SAA, protein akutní fáze), amyloidóza vzniká u chronických zánětů.
- ATTR – nejčastěji díky mutaci pro transtyretin (TTR, transportní protein pro hormony štítné žlázy), nejčastější familiární forma.
- Aβ2M – β2mikroglobulin, amyloidóza vzniká u pacientů s dlouhodobým chronickým renálním selháním léčených dialýzou.
- Aβ – nejčastější forma lokalizované amyloidózy, depozita přítomna v mozku u pacientů s Alzheimerovou demencí.
Diagnostika
- K identifikaci byla dříve prováděna biopsie gingivální a rektální sliznice, v současnosti lze provést biopsii břišního tuku (aspirace 16G jehlou v lokální anestezii).
- Pokud je vyšetření negativní, je vhodné provést biopsii jazyka, bukální sliznice nebo rekta.
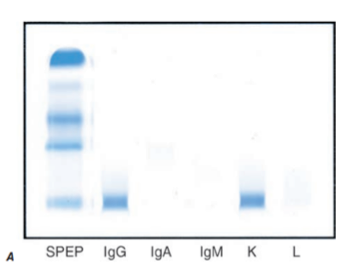
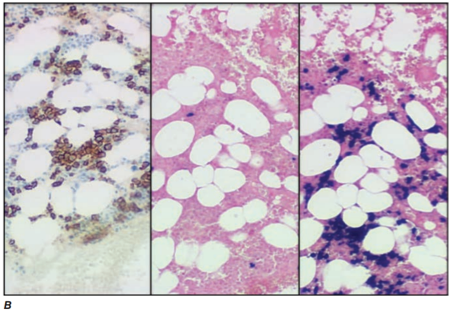
- V případě negativity nutné provést invazivní biopsii postiženého orgánu (ledviny, srdce, játra, GIT apod.). Materiál je potřeba nabarvit Kongo červení, dále lze prokázat zelený dvojlom v polarizačním mikroskopu nebo přímo prokázat 10 nm amyloidové fibrily elektronovým mikroskopem.
- Po identifikaci amyloidózy je imunohistochemicky, imunoelektronmikroskopicky nebo spektrometricky identifikován konkrétní typ amyloidu. V případě podezření na familiární formu je indikováno genetické vyšetření.
AL amyloidóza
Epidemiologie – incidence je 1 : 20000 ročně, většinou u jedinců > 40 let, nejčastěji starších než 60 let.
Etiologie a patofyziologie – nejčastěji způsobena klonální expanzí plazmocytů, které produkují monoklonální lehké řetězce. Ty buď dávají vznik mnohočetnému myelomu nebo se defektně skládají se vznikem AL amyloidózy. AL amyloidza vzniká nejčastěji při mnohočetném myelomu nebo B lymfoproliferací (non-Hodgkinské lymfomy nebo Waldenströmova makroglobulinémie). Amyloidové fibrily jsou tvořeny volnými lehkými řetězci nebo jejich fragmenty, častěji podtypem lambda.
Klinický obraz
Časté jsou nespecifické příznaky jako váhový úbytek a únava. Nejčastějším postiženým orgánem jsou:
- ledviny (v 70 – 80 %) – sekundární glomerulonefritida s nefrotickým syndromem (proteinurie, hypoalbuminémie, otoky, hypercholesterolémie, tendence k trombózám).
- srdce (v 50 – 60 %) – nejčastější příčinou smrti, na EKG je nízká voltáž v končetinových svodech, echokardiograficky koncentrická hypertrofie, výrazná diastolická dysfunkce, „jiskřivý“ vzhled myokardu je spíše popisovan v literatuře a dále dilatace levé síně, na MRI je ve fázi pozdního sycení (LGE) poměrně heterogenní nález (sycení subendokardiální, midmyokardiální apod), běžné je zvýšení NTproBNP.
- periferní senzomotorická a vegetativní neuropatie s autonomní dysfunkcí vede k poruchám gastrointestinální motility (časná sytost, průjem nebo zácpa), suché oči, nedostatek slin, erektilní dysfunkce, ortostatická hypotenze nebo neurogenní měchýř.
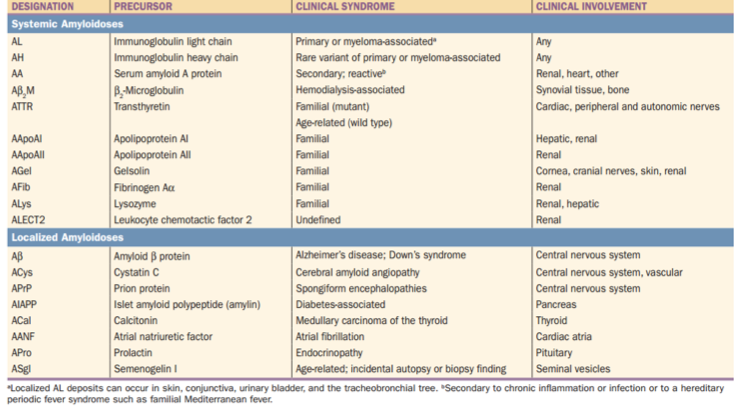
- makroglosie (lze prokázat u cca 10 % pacientů), poměrně patognomonická
- játra se vznikem cholestázy a hepatomegalie
- tendence ke vzniku sufúzí – amyloid se hromadí v kapilárách a zároveň vede k vyvazování faktoru X. Sufúze vznikají nejčastěji v okolí očí, kde vytváří „znamení mývala“
- Mezi ostatní známky patří dystrofie nehtů, alopecie, amyloidová artropatie a tendence ke vzniku syndromu karpálního tunelu.
Diagnostika
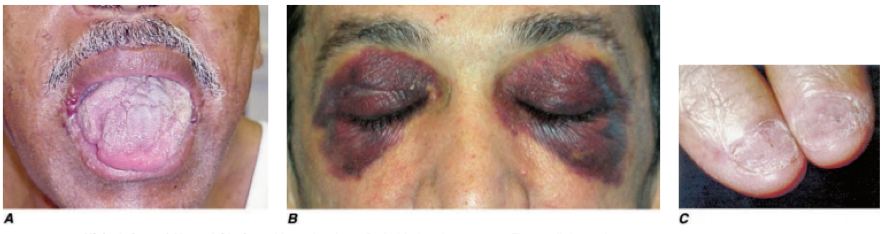
- Základní znakem cílem je identifikovat B-lymfoproliferativní proces jako zdroj klonálních lehkých řetězců u amyloidózy. Samotná elektroforéza bílkovin séra je nedostatečná (citlivost metody je nedostatečná, protože i již klinicky signifikantní množství nemusí vykreslit typický „M spike“ v séru nebo moči (Bence-Jonesova bílkovina).
- U 90 % pacientů lze volné lehké řetězce nebo imunoglobuliny v séru a moči detekovat pomocí imunofixační elektoforézy. Důležité je vyšetření absolutního množství i poměru κ/λ (při chronické renální insuficienci se nespecificky snižuje clearance obou typu volných lehkých řetězců a roste jejich hladina). Jejich monoklonalitu lze prokázat flow cytometrií, imunohistochemicky, nebo in situ hybridizací.
Terapie – přežití neléčené amyloidózy je 1 – 2 roky od jejího stanovení, v případě srdeční amyloidózy cca 6 měsíců. Léčba:
- Hematologická – vysokodávkový melfalan + dexametazon + autologní transplantace kmenových buněk (HDM/SCT) navodí kompletní remisi u cca 40 %. Imunomodulátory thalidomid, lenalidomid a pomalidomid, dále inhibitor proteázomu bortezomib. 6 – 12 měsíců po dosažení hematologické odpovědi lze prokázat zlepšení funkce postižených orgánů.
- Srdce – často indikována transplantace srdce. CAVE Kontraindikován digoxin, protože se váže na amyloidové fibrily a může nadměrně zvyšovat svoji koncentraci. Implantace ICD má sníženou efektivitu vzhledem ke ztluštění myokardu.
- Ledviny – diuretika s albuminem. ACEI nezpomalují progresi nefrotického syndromu.
- Autonomní neuropatie – hypotenzi lze léčit alfa agonisty (midodrin), porucha motilility prokinetiky a vlákninou.
- Lokalizované amyloidóza (dýchací cesty, močový měchýř, kůže a lymfatické uzliny) – chirurgická léčba nebo nízkodávková radioterapie (20 Gy).
AA amyloidóza
Její příčinou je chronicky zvýšená hladina SAA (serum amyloid A), která se objevuje při dlouhotrvajících infekcích a a zánětech (revmatoidní artritida, idiopatické střevní záněty apod.). Vzhledem ke zlepšení protizánětlivé léčby její prevalence klesá (< 2 % osob s těmito chorobami). Jako první jsou postiženy ledviny, objevit se mohou i hepatomegalie, splenomegalie a autonomní neuropatie, v pozdějších fázích se objevuje i kardiomyopatie (nelze klinicky rozlišit od AL amyloidózy). Léčbou je vyřešení vyvolávající choroby.
ATTR a AF amyloidóza
Epidemiologie a etiologie – klinické projevy většinou začínají ve středním věku, jde o vzácnou chorobu s incidencí < 1:100000.
Klasifikace – na základě typu vyvolávajícího proteinu:
- ATTRm – mutovaným transtyretinem (TTR, prealbumin, známo 120 typů mutací)
- ATTRwt – nemutovaným transtyretinem (wild type), spojena se stárnutím (dříve senilní amyloidóza). Velmi často zjištěna u pacientů s amyloidovou kardiomyopatií ve věku > 65 let, při pitvách pacientů > 80 let byla nalezena depozita amyloidu ve 25 % srdcí.
- ostatní (AI, AII, gelsolin, fibrinogen Aα, lysozym) jsou raritní
Patogeneze – kritickým momentem je disociace TTR tetramerů do monomerů s jejich následnou defektní polymerizací.
Klinický obraz – klinický obraz je u různých forem variabilní v závislosti na mutovaném proteinu. ATTR se projevuje ascendentní polyneuropatií (nejdříve dolních, poté horních končetin), včetně autonomní (ortostatická hypotenze, průjem a váhový úbytek) a postižením srdce.
U ostatních forem je typické postižení ledvin (fibrinogen, lysozym, apolipoproteiny), jaterní (apo A1), postižení hlavových nervů a rohovky (gelsolin).
Léčbou volby je transplantace jater, která ale nezvrátí polyneuropatii a je proto nejúspěšnější v počátečních stádiích. Doba přežití bez léčby je 5 – 15 let. Stabilizace TTR tetramerů probíhá buď vazbou tyroxinu nebo „thyroxin mimetiky“, které stabilizují TTR tetramery (diflunisal, tafamidis). Nejnovějším přístupem je genová terapie s utlumením exprese genu pro TTR a jeho produkci játry.
Aβ2M amyloidóza
Původem amyloidu je beta2-mikroglobulin, který má při chronickém selhání ledvin sníženou clearance, hromadí se a zároveň není konvenční hemodialýzou odstraňován. Projevuje se revmatologickou symptomatologií (syndrom karpálního tunelu, bilaterální postižení velkých kloubů s výpotkem). Synoviální tekutina je nezánětlivá a při barvení sedimentu Kongo červení lze prokázat amyloid. Specifická terapie neexistuje, řešením je přerušení hemodialýzy díky úspěšné transplantaci ledvin.