Epidemiologie – incidence zánětlivých myopatií (IM) je 1:25000 s prevalencí 1:5000, ženy jsou postiženy 3x častěji než muži (myozitida s inkluzními tělísky je naopak častější u starších mužů), maximum věku výskytu je 50 – 60 let (vyjímkou ale nejsou ani i juvenilní myopatie).
Etiologie – není známá
- Genetická predispozice (HLA-DRB103, DQA1*05-DQB1*02)
- Vnější faktory – virové infekce, UV záření (dermatomyozitidy), léky (penicilamin)
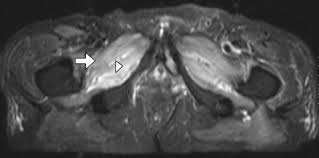
Obecný diagnostický přístup – při podezření na IM je základem lokalizace postižených svalových skupin dále snaha si všimnout, zda je přítomno kožní postižení (heliotropní rash, Gottronovy papuly a dilatace kapilár nehtového lůžka). Dále je vhodný odběr kreatin kinázy (CK), při lokalizaci léze je vhodné EMG a MRI a definitivní diagnózu dá svalová biopsie.
Klasifikace
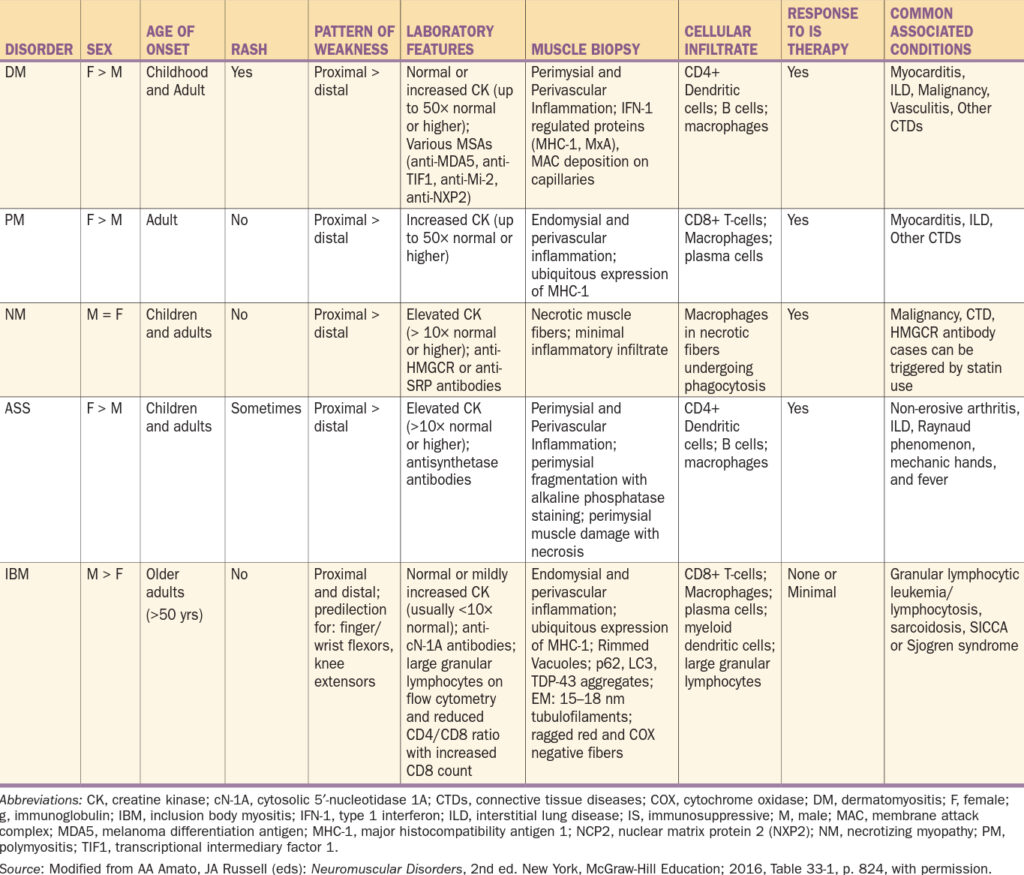
I. Dermatomyozitida
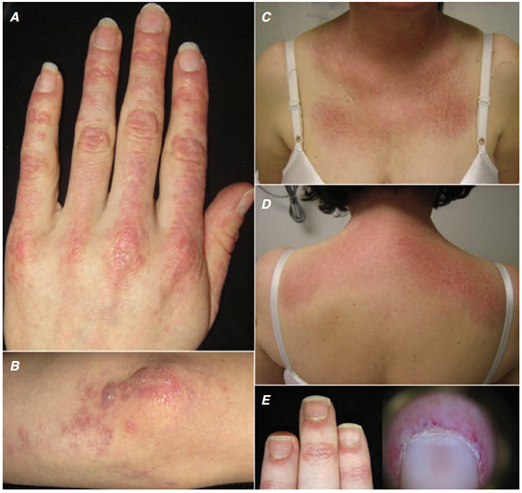
Postihuje proximální svalové skupiny s charakteristickým kožním postižením:
- Otok očí, zejména horních víček
- Červenofialové zbarvení kůže čela, tváří, krku, hrudníku (zde ve tvaru V), na zádech, loktech a vnitřních kotnících (vystavených Slunci)…heliotropní exantém.
- Tmavě červený až fialový, mírně vyvýšený nesvědící exantém na extenzorové straně nad drobnými klouby ruky…Gottronovy známky.
- Hyperémie a teleangiektázie nehtových lůžek
U dermatomyozitidy se soudilo, že protilátky a komplement vedou k destrukci endotelu s následnou ischemizací. Dle posledních studií je pravděpodobné, že hlavním mediátorem je IFN- β. Dochází k perimyziální infiltraci svalu CD4+ lymfocyty, makrofágy a dendritickými buňkami. Další klinické projevy odpovídají ostatním IM.
CAVE Dermatomyozitidy mají v 15 % asociaci s malignitami.
II. Polymyozitida
Heterogenní skupina chorob spojená s proximální svalovou slabostí. I zde je častá asociace s malignitami, i když menší než při dermatomyozitidách. V biopsii je častější perimyziální (může být i endomyziální) infiltrace CD8+ lymfocyty bez známek nekrózy svalstva.
Overlap syndromy – IM se mohou překrývat se sklerodermií, smíšenými chorobami pojiva, systémovým lupus erthematodes nebo revmatoidní artritidou.
III. Antisystetázový syndrom
= horečka + intersticiální postižení plic + neerozivní artritida + myozitida + Raynaudův fenomén + prsty mechanika (zhrubění a olupování kůže na radiální straně) + protilátky proti aminoacyl – tRNA syntetáze (anti-Jo-1)
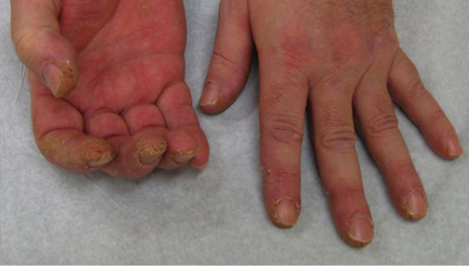
Protilátky proti aminoacyl-tRNA syntetáze (z nichž nejčastější je anti-Jo-1) jsou přítomny u cca 30 % pacientů s myozitidou. Příznivé je, že antisysntetázový syndrom nebývá spojen s vyšší incidencí malignity.Histologicky lze nalézt perimyziální poškození a fragmentaci (větší než u dermatomyozitidy) s infiltrací CD8 lymfocyty.
IV. Myozitida s inkluzními tělísky
Většinou postihuje jedince nad 50 let věku (lehce častěji muže, než ženy). Na rozdíl od polymyozitidy je svalová slabost často asymetrická, často je postiženo předloktí (flexor digitorum profundus) a svalstvo stehen (vastus medialis a lateralis). Hladina kreatinkinázy je jen mírně elevovaná (max do 10 násobku normy a poměrně specifická je pozitivita anti-cN-1A (protilátky proti cytozolové 5′-nukleotidáze 1A), dále je typický snížený poměr CD4/CD8 lymfocytů se zvýšeným počtem CD8. Typická je špatná odpověď na léčbu. Charakteristickým znakem je přítomnost inkluzních tělísek obsahujících amyloid. Choroba pacienty většinou po 10 – 15 letech upoutá na vozík.
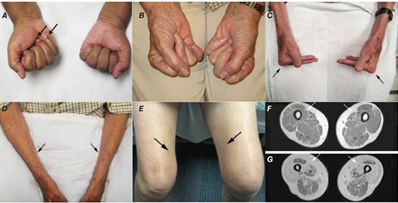
Klinický obraz IM – v předchorobí lze často vypátrat horečnaté onemocnění, mezi prodromy patří artritida nebo Raynaudův fenomén.
- I. Svaly – svalová slabost postihuje proximální svalové skupiny končetin, trupu a krku. Následně vzniká atrofie, která klinicky nemusí být patrná (svaly jsou nahrazeny tukem a vazivem). Postupně mohou vznikat kontraktury. Bolest není nutným symptomem choroby. Projevy postižení:
- dolních končetin – obtíže při chůzi do schodů
- horních končetin – potížemi při zvedání předmětů
- svalstva jícnu – dysfágie s rizikem aspirace
- svalstva hrtanu a měkkého patra – chrapot, nazolálie
- II. Klouby – neerozivní artralgie
- III. Plíce – intersticiální plicní fibróza, riziko aspirace (prokazatelné na HRCT).
- IV. Srdce – na EKG zejména poruchy převodu a arytmie (LAH, RBBB, SA blokády, síňové a komorové extrasystoly), jen vzácně srdeční selhání.
- V. Kalcifikace – kalcifikace měkkých tkání je častější u dětí, v dospělosti vzácné.
Diagnostika
- 1. Laboratorní testy – výrazné zvýšení CK a myoglobinu, nespecificky nespecificky zvýšení LDH, AST. Dále normální FW a CRP. Protilátky:
- antisyntetázové autoprotilátky (anti-Jo-1)
- anti-PM-Scl – specifické pro polymyozitidu
- anti-Mi-2 – specifické pro dermatomyozitidu
- 2. EMG – ukazuje myopatické postižení
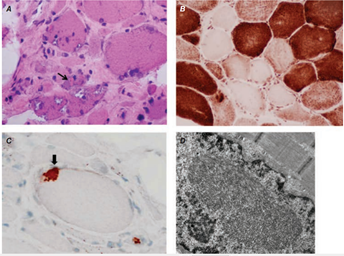
- 3. Biopsie – zánětlivý infiltrát, atrofie svalových vláken, případně jejich regenerace. Dermatomyozitida – perifascikulární atrofie a perivaskulární infiltrace CD4+ T- lymfocyty, makrofágy a B-lymfocyty. Polymyozitida – aktivované CD8+ T-lymfocyty, které obklopují a invadují neznekrotizovaná svalová vlákna.
- 4. MRI postižených svalů prokazuje edém.
Diagnostická kritéria
- Proximální, symetrická svalová slabost, progredující po týdny nebo měsíce (s/bez přítomné bolesti, s/bez kožních změn).
- Elevace CK a myoglobinu
- Patologické EMG
- Bioptický průkaz myozitidy
Terapie
- 1. volba glukokortikoidy – prednison v dávce 1 mg/kg/den po dobu 4 – 6 týdnů s postupnou detrakcí a dobou podávání nejméně 1 rok. Při vysoké aktivitě nemoci (dysfagie) nutné zahájit pulsy metylprednisolonu.
- 2. volba imunosupresiva při život ohrožujícím průběhu nebo nedostatečné odpovědi na kortikoidy (cyklofosfamid, cyklosporin, takrolimus, mykofenolát mofetil, methotrexát, azathioprin).
- Kalcifikace se mohou provalit navenek nebo chirurgicky extirpovat.
Prognóza
- Děti – 50 % kompletní remise, 35 % pacientů vyžaduje protrahovanou léčbu, 15 % jsou těžké, život ohrožující stavy.
- Dospělí – horší prognóza (zejména u paraneoplastických myozitid). Většinou nutná dlouhodobá léčba. Čím časnější stanovení diagnózy, tím lépe.