Aplastická anémie
Aplastická anémie vzniká v důsledku poškození kmenových buněk krvetvorby (CD34+). Roční incidence v České republice je 1 : 500 tisíc (v Asii až 3x vyšší). Těžká forma je definována jako pokles buněčnosti kostní dřeně < 30 % + dva ze tří:
- retikulocyty < 0,1 %
- neutrofily < 0,5 × 109/l
- trombocyty < 20 × 109/l
U 2/3 pacientů se nezjistí příčina, u zbytku je jako příčina identifikována:
- virová infekce (EBV, parvovirus B19, hepatitidy, HIV).
- léky (zlato, NSAID, sulfonamidy – idiosynkraticky nebo přímo myelotoxicky po podání cytostatik).
- chemické látky (benzen).
- radioaktivní záření (obecně poškozuje zejména tkáně s vysokým obratem).

Vlastním mechanismem je ve velké většině autoimunitní reakce proti kmenovým buňkám pomocí cytotoxických T lymfocytů, v případě radioaktivního záření přímé poškození kmenových buněk, některé viry se navíc umí inkorporovat do DNA. Získaná aplastická anémie vzniká často u dříve zdravých mladých jedinců, často ve spojení s neinfekční hepatitidou. Je příbuzná s paroxysmální noční hemoglobinurií a myelodysplastickým syndromem, mezi těmito chorobami je často neostrý přechod.
Většinou se projevuje kombinací příznaků trombocytopenie (krvácení), neutropenie (infekce) a anémie (dušnost, únava).
Diagnóza je dána nálezem v periferní krvi a potvrzena průkazem hypoplastické dřeně s tukovými oky, bez známek fibrózy, infiltrace nebo myelodysplazie. Typický je velmi nízký počet retikulocytů.
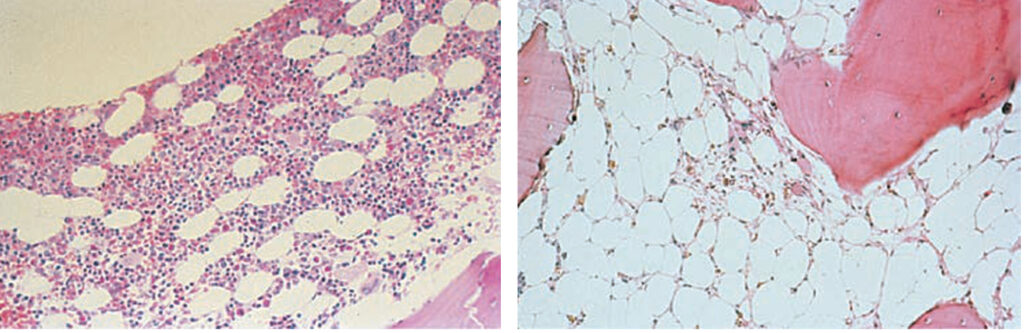
Léčbu lze provádět několika způsoby:
- náhrada hematopoetických buněk pomocí alogenní transplantace kostní dřeně (SCT), což je optimální u mladých jedinců.
- u starších jedinců suprese buněk, tak aby měla možnost regenerovat normální hematopoéza – antithymocytární globulin + cyklosporin A, často v kombinaci s eltrombopagem (syntetický analog trombopoetinu, který má nečekaný účinek na zvýšení proliferace všech tří řad).
Zásadní je podpůrná léčba (transfúze erytrocytů a trombocytů, včasné podání širokospektrých antibiotik při infekci a růstových faktorů). Pozdní komplikací je vznik MDS s možným přechodem do leukémie.
Čistá aplázie červené krevní řady
Aplázie erytrocytární řady (PRCA) při vynechání ostatních buněčných linií. Typickým nálezem je anémie i nedostatek retikulocytů a erytrocytárních prekurzorů. Etiologicky:
- Idiopatická PRCA (Diamond – Blackfanův syndrom) – AR dědičná PRCA, mimo anémii je přítomna porucha růstu i mentálního vývoje, hypoplázie ledvin a anomálie palce (tříčlánkový). Základem léčby je imunosupresivní (glukokortikoidy, cyklosporin, ATG, azathioprin, cyklofosfamid) a symptomatická terapie (transfúze, chelatační léčba).
- Infekce parvovirem B19 – za normálních okolností dojde ke vzniku neutralizačních protilátek s rychlým ústupem anémie, ale u imunosuprimovaných může dojít k chronické infekci i anémii. V tomto případě je dobrá reakce na IVIG.
- Thymom, vzácneji lymfoproliferace nebo tumory, léčbou je resekce nebo jiná specifická léčba.
- Autoprotilátky proti EPO
- Léky (fenytoin, azathioprin, izoniazid) – zásadní je vysazení léčby.
Kongenitální dyserytropoetická dysplázie
Kongenitální dysertropoetické anémie (CDA) je velmi vzácná, patogeneticky poměrně heterogenní skupina vrozených poruch antigenní výbavy erytrocytů.
- CDA I. typu – AR dědičná megaloblastová přestavba kostní dřeně s vícejaderými erytroblasty, která se projevuje středně těžkou až těžkou anémií. Léčebně se podává IFN-α.
- CDA II. typu (HEMPAS – hereditary erythroblastic multinuclearity with positive acidified serum) – AR dědičná megaloblastová přestavba kostní dřeně s vícejaderými erytroblasty, u kterých je přítomen fenomén dvojité membrány. Zlepšení po splenektomii, těžké formy indikovány k SCT.
- CDA III. typu – kostní dřen s gigantoblasty, většinou středně významná anémie s bazofilním tečkováním erytrocytů v periferní krvi.
Vzácně vrozené poruchy kmenové buňky
Fanconiho anémie – vzácná AR dědičná choroba (prevalence 1:130 tisíc, častěji u Askenázských Židů). Většinou se projeví do 7 let dítěte. Příčinou jsou mutace genů, které se účastní reparace DNA. Je přítomna vrozená hypoplazie kostní dřeně s pancytopenií (včetně makrocytární anémie) v krevním obraze, dále vývojové poruchy (nízká výška, typický obličej – široký kořen nosu, epikanty, mikrognatie, chybění radia, abnormální palce, hypogonadismus u mužů, hypoplázie očí a ledvin), hluchota, hnědé kožní pigmentace a sklon k maligní transformaci (nejčastěji MDS a leukémie). Typickým laboratorním nálezem je zvýšené množství HbF. Diagnóza je dána testem stability chromozómů a průtokovou cytometrií. První linií terapie je podávání androgenů a růstových faktorů (odpověď u 50 – 70 % pacientů), jedinou kurativní metodou je SCT.
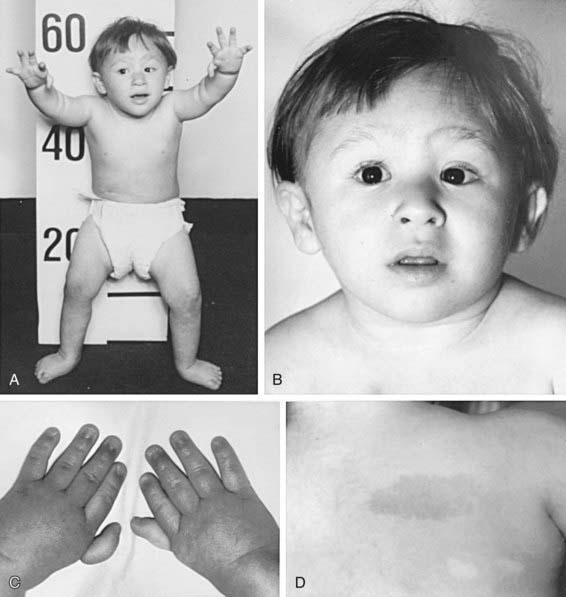
Dyskeratosis congenita – onemocnění způsobené předčasnou redukcí délky telomer s následnou omezenou proliferaci kmenových buněk. Typická je tetráda: aplastická anémie + hyperpigmentace + leukoplakie + dystrofie nehtů.

Shwachman – Diamondův syndrom – projevuje se časně neutropenií a pankreatickou insuficiencí s malabsorpcí a retardací růstu. Léčbou volby je SCT.
Myelodysplastický syndrom
Definice – myelodysplastický syndrom (MDS) je klonální onemocnění krvetvorby, při kterém vzniká iniciální poškození genomu kmenové buňky s následnou tvorbou patologického klonu. Dochází ke stimulaci proliferace časných a forem krvetvorby se zvýšeným stupněm apoptózy, což v počátečních fázích choroby vede k obrazu dysplastické kostní dřeně bohaté na buňky a cytopenii v periferní krvi. Klony mají nestabilní genom s následnými mutacemi, které vedou ke ztrátě autoregulace, postupnému nárůstu blastů a přechodu do akutní myeloidní leukémie.
Klasifikace – původní byla FAB, poté se vyvíjela WHO, nyní se používá poslední verze z roku 2016.
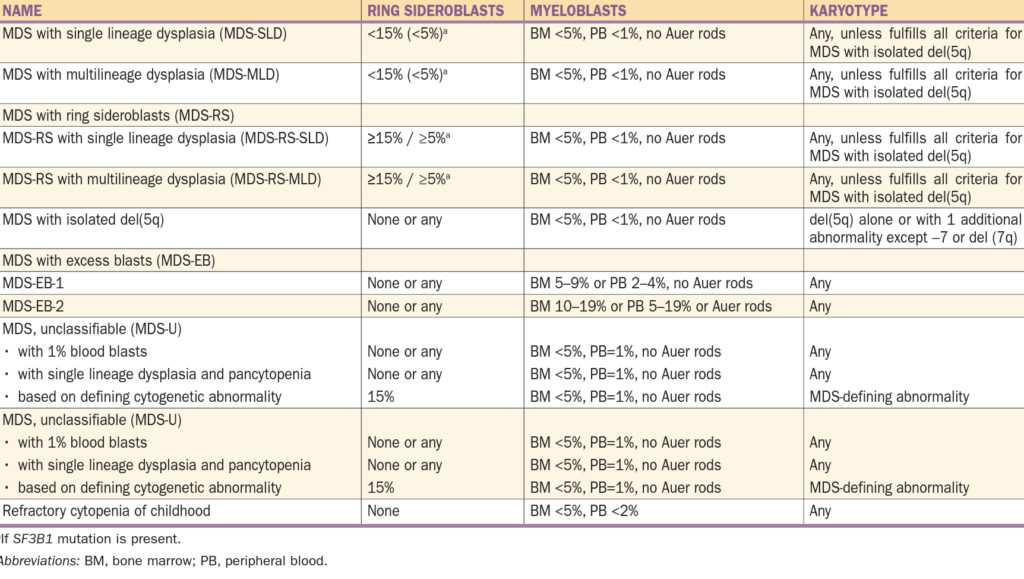
Zjednodušeně:
- MDS s dysplazií v jedné řadě / ve více řadách (kostní dřeň (KD) < 15 % prstenčitých sideroblastů, periferní krev (PK) < 5 % myeloblastů)
- MDS s prstenčitými sideroblasty s dysplazií v jedné řadě / ve více řadách (KD > 15 % prstenčitých sideroblastů). Prstenec je tvořen mitochondriemi obsahujícími nadbytek železa ve formě feritinu.
- MDS s izolovanou delecí 5q
- MDS s nadbytkem blastů (KD 5 – 9 % myeloblastů – MDS EB-1, 10 – 19 % myeloblastů MDS EB-2)
- MDS neklasifikovaný
Epidemiologie – MDS je choroba starších lidí (zde i 1 : 2000), u mladých je velmi vzácný a téměř vždy se zde jedná o genetickou odchylku).
Etiologie a patofyziologie – MDS je často spojen s expozicí radioaktivního záření, benzenu a stavu po chemoterapii (alkylační cytostatika po 5 – 7 letech od podání, inhibitory topoizomerázy po cca 2 letech od podání). Typický pacient s MDS ale nemá jasnou anamnézu těchto faktorů.
V současnosti bylo identifikováno již > 100 genů, které jejichž mutace působí MDS (řada z nich je současně spojována i s akutní myeloidní leukémií).
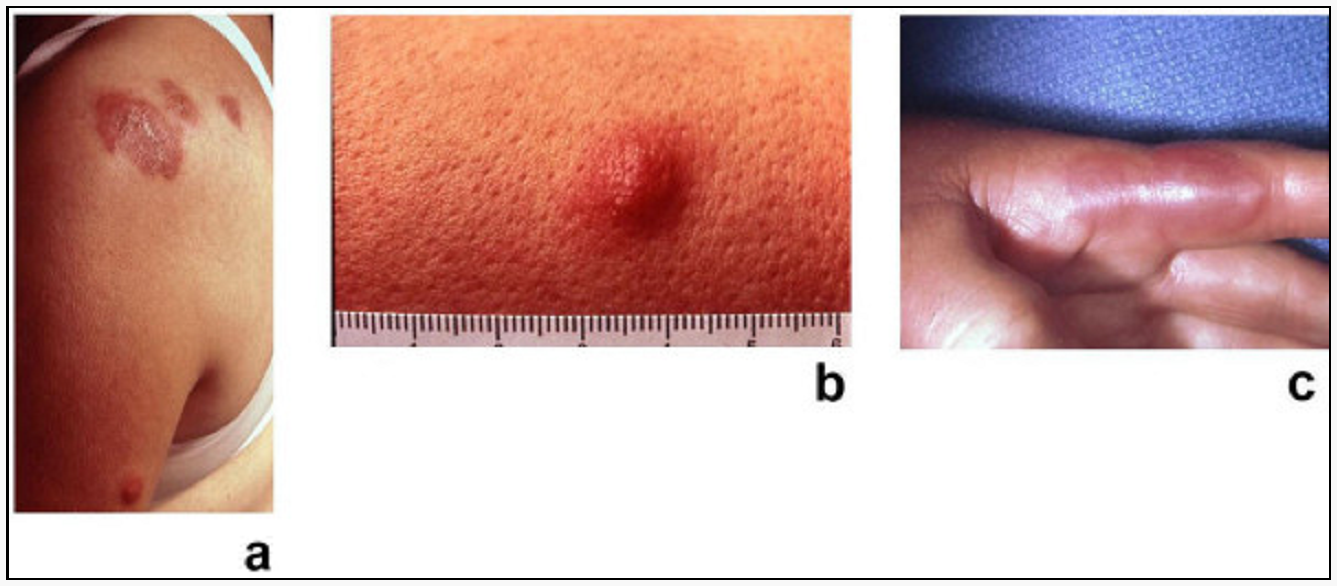
Klinický obraz – v klinickém obraze dominuje zpočátku anémie, horečka a váhový úbytek není typický (spíše by měl upozornit na myeloproliferaci než myelodysplázii), 20 % pacientů má splenomegalii, u některých se může objevit kožní postižení (např. Sweetův syndrom – erythematózní nebo lividní uzly v kůži odpovídající neutrofilní infiltraci).
Klasifikační schémata zároveň i stratifikují nemocné podle prognózy. Obecně platí, že nemocní bez zmožení blastů mají lepší prognózu než nemocní s počtem blastů v kostní dřeňi nad 5%, kteří mají častější přechod do akutní myeloidní leukemie. Na základě těchto zjištění bylo vypracováno IPSS-R skóre: https://www.mds-foundation.org/ipss-r-calculator/ (posuzuje karyotyp, počet blastů v kostní dřeni, hemoglobin, trombocyty, neutrofily). Většina pacientů nezemře na leukémii, ale na komplikace spojené s pancytopenií.
Diagnostika – základním kritériem je přítomnost nespecifických dysplastických změn v kostní dřeni u > 10 % buněk dané vývojové řady.
- periferní krev – typickým nálezem je anémie (často makrocytární), někdy pancytopenie. Vážným prognostickým nálezem jsou cirkulující blasty.
- kostní dřeň – normální, hypercelulární, ale ve 20 % i hypocelulární. Přítomny dyserytropoetické a jiné nespecifické změny erytrocytů i ostatních vývojových řad > 10 % buněk. Cytogenetické vyšetření může identifikovat chromozomální abnormalitu. Pokud je počet blastů > 20 % jde již o akutní myeloidní leukémii. Je vhodné zhodnotit i buněčnost dřeně a její fibrózu.
Terapie
- jedinou léčebnou metodou je alogenní transplantace kostní dřeně.
- u hypoplastické formy lze použít kortikosteroidy, cyklosporin, ATG a hypometylační látky (azacitidin, decitabin).
- při nálezu prstenčitých sideroblastů lze diagnosticko – terapeuticky vyzkoušet pyridoxin.
- Při 5q- lenalidomid.
- symptomaticky lze používat tranfúze erytrocytů, trombocytů a použití chelatačních látek při přetížení železem, dále erytropoetin a G-CSF.