Do skupiny Ph- myeloproliferací patří celkem 8 chorob (některé velmi vzácné), které se vyznačují nepřítomností translokace t[9;22] známé jako Filadelfský chromozóm. Naopak, častá je mutace tyrozin kinázy JAK2.
Polycythemia vera
Definice – polycythemia vera (PV) je chronické myeloproliferativní onemocnění, při kterém dochází ke zvýšené proliferaci erytrocytů. Incidence je 1-2:100 tisíc a stoupá s věkem. Příčinou většiny případů je mutace JAK2.
Gen pro JAK2 je za běžných okolností aktivován erytropoetinem, cestou jeho receptoru. Vzniklá tyrozinkináza stimuluje proliferaci a diferenciaci erytrocytů a tlumí apoptózu. Současně je stimulována i leuko- a trombopoéza.
Klinický obraz – průběh má 3 fáze:
- prepolycytemická – mírná erytrocytóza
- polycytemická – signifikantní zmnožení krevní masy
- postpolycytemická – stádium myelofibrózy, pancytopenie a extramedulární hematopoézy se splenomegalií. Terminálně může přecházet do akutní leukémie.
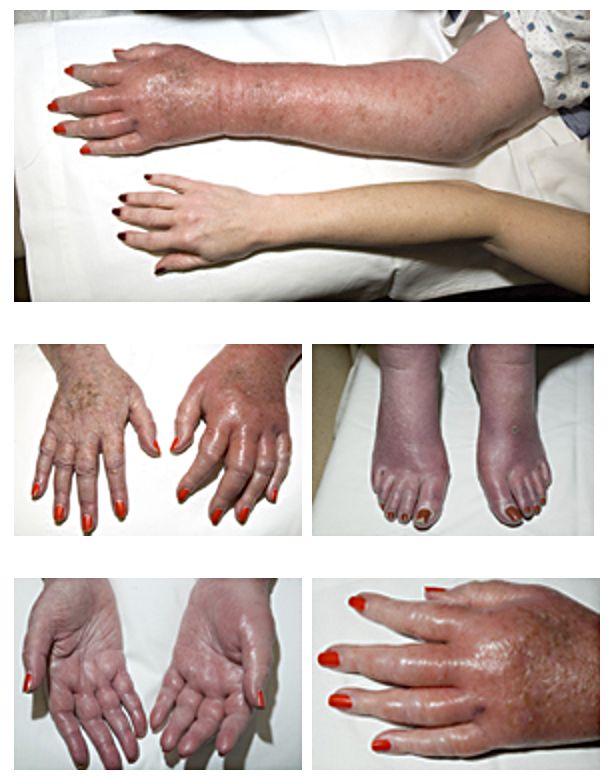
Začátek choroby je nespecifický (zvýšená únavnost, hubnutí, noční pocení), postupně vznikají příznaky plynoucí ze zmnožení množství erytrocytů, např. bolesti hlavy, závratě, dušnost, poruchy zraku, pruritus po teplé koupeli a erytromelalgie. Typická je pletora s překrvením spojivek, častá je splenomegalie a hepatomegalie.
Se stoupajícím věkem roste riziko trombotických komplikací, naopak, při počtech trombocytů > 1000 je vysoké riziko krvácení (spotřeba multimerů vWF vysokým počtem trombocytů).
Diagnostika – v současnosti je doporučeno řídit se diagnostickými kritérii dle WHO (2008). Pro diagnózu musí být splněna dvě velká a jedno malé nebo jedno velké a dvě malá kritéria:
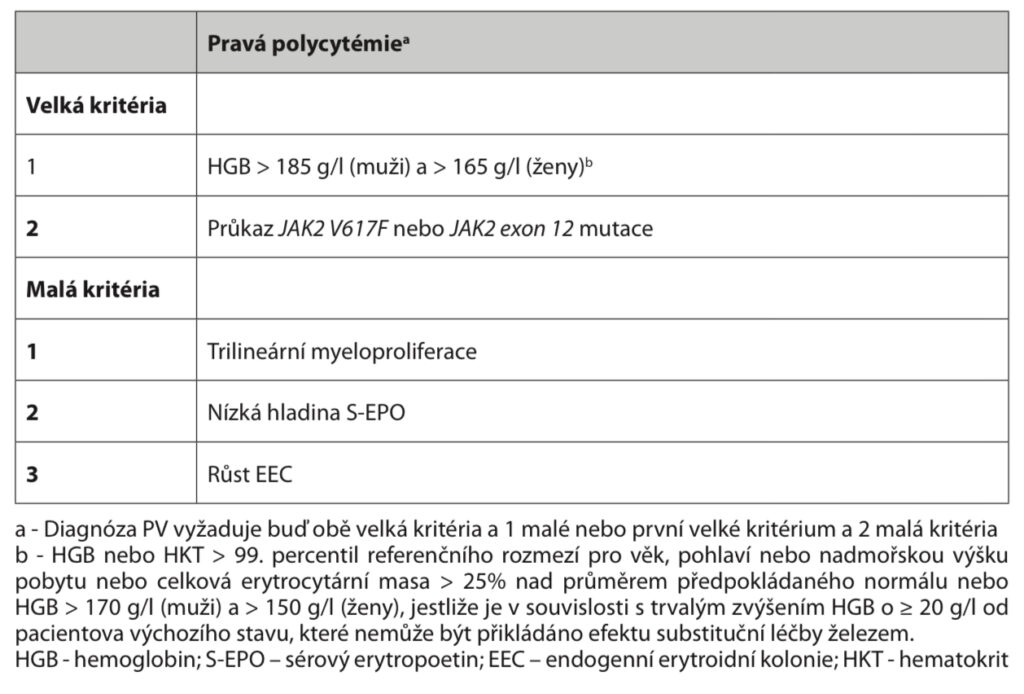
Typické je zvýšení hemoglobinu (> 185 g/l u mužů, > 165 g/l u žen), leukocyty bývají také zvýšené (většinou < 20), trombocyty jsou normální nebo lehce zvýšené. Hladina EPO je snížená a je přítomen autonomní růst endogenních erytroidních kolonií (EEC). Častá je splenomegalie, základem je průkaz mutace JAK2, většinou v pozici V617F. Kostní dřeň je hypercelulární, při přechodu do fibrotické fáze je hypocelulární a firbotická.
CAVE Je nutné vyloučit sekundární polycytémii, která bývá z těchto příčin:
- následek celkové hypoxie (onemocnění plic, cyanotické srdeční vady, AV zkraty, hemoglobinopatie, methemoglobinéme, karboxyhemoglobinémie, pobyt ve vysoké nadmořské výšce)
- hypoxie ledvin (polycystické ledviny, stenóza a. renalis)
- paraneoplasticky (Grawitzův tumor, hepatocelulární karcinom, feochromocytom)
- otrava kovy (kobalt)
Terapie – dle stupně rizika:
- nízké riziko – ASA v nízké dávce + venepunkce (cílový hematokrit < 0,45)
- vysoké riziko – + navíc cytoredukční léčba hydroxyureou a/nebo interferonem α, při jejich intoleranci ruxolitinib (inhibitor JAK1 a JAK2).
U nestabilních pacientů nebo při přípravě na operaci lze použít erytrocyt – nebo trombocytaferézu při extrémní trombocytóze (> 1500 – 2000), při které je i třeba hodně zvažovat indikaci antiagregace vzhledem k získané von Willebrandově chorobě. Při trombocytóze > 1000 je ke zvážení anagrelid.
Esenciální trombocytémie
Definice – esenciální trombocytémie (ET) je chronická myeloproliferace, která se vyznačuje trombocytózou (> 450) se zvýšeným množstvím megakaryocytů v kostní dření a sklonem ke krvácení nebo trombózám. Incidence je 1-2:100 tisíc. Etiologie choroby není jasná (u 50 % je přítomna mutace JAK2).
Klinický obraz – polovina nemocných je asymptomatická, u poloviny nemocných jsou typická krvácení nebo trombózy (erytromelalgie, migrény, TIA). Onemocnění může přecházet do myelofibrózy, ale již ne do leukémie.
Diagnostika – dle WHO je nutné splnění čtyř kritérií:

V krevním obraze je vždy trombocytóza (> 450) a normální nebo lehce zvýšený počet leukocytů (< 15). V periferní krvi nejsou na rozdíl od myelofibrózy přítomny mladší nezralé prekurzory (blasty). Geneticky lze vyšetřit mutaci JAK2 V617F (odlišení od sekundární trombocytózy), BCR-ABL je negativní.
Terapie – cílem léčby je prevence trombóz a zmírnění symptomů. Intenzita terapie je dle stupně rizika:
- nízké riziko – ASA v nízké dávce
- vysoké riziko – + navíc cytoredukční léčba hydroxyureou, při rezistenci anagrelid nebo interferon α.
U vysokého rizka krvácení při extrémní trombocytóze (> 1500 – 2000) lze použít trombocytaferézu a je i třeba hodně zvažovat indikaci antiagregace vzhledem k získané von Willebrandově chorobě.
Primární myelofibróza
Definice – primární myelofibróza je chronická myeloproliferace (zeména megakaryocytární a granulocytární řady) s progresivní fibrotizací kostní dřeně a následným rozvojem extramedulární hematopoézy. Incidence je cca 1:100 tisíc.
Etiologie – není jednoznačná. Popsány familiární formy a dále vznik po expozici benzenu a ionizačního záření. Fibróza vzniká sekundárně jako reakce na myeloproliferaci.
Klinický obraz – choroba má tři fáze:
- prefibrotická – různě dlouhá, typická je hypercelulární kostní dřeň. V periferním KO je častá leukocytóza a trombocytóza.
- fibrotická – výrazná fibróza kostní dřeně. Splenomegalie odráží stupeň fibrózy kostní dřeně. Při vyšetření kostní dřeně je nález zmnožení granulocytární a trombocytární řad a jen mírnáí fibróza.
- osteosklerotická – terminální obraz, kdy nefungue kostní dřeň se vznikem hepatosplenomegalie a blasty v periferním krevním obraze. Přítomna je pancytopenie a přítomnost CD34+ buněk a blastů v periferní krvi. Při trepanobiopsii je typická pokročilá fibróza až osteoskleróza.
CAVE Ve 20 % dochází k rozvoji akutní myeloidní leukémie.
30 % nemocných je v době stanovení diagnózy asymptomatických a je zjištěno náhodně při došetření trombocytózy nebo splenomegalie (u 90 % nemocných) a/nebo hepatomegalie (u 50 % nemocných). Přítomny jsou i nespecifické příznaky (váhový úbytek, subfebrilie, únava, noční poty, pruritus, dna…).
Diagnostika – nález v kostní dření a krevním obrazu viz výše. Může být přítomna mutace JAK2 V617F (vzácněji MPL nebo CARL). Ke stanovení diagnózy musí být splněna všechna tři velká kritéria a alespoň dvě malá kritéria WHO 2008.
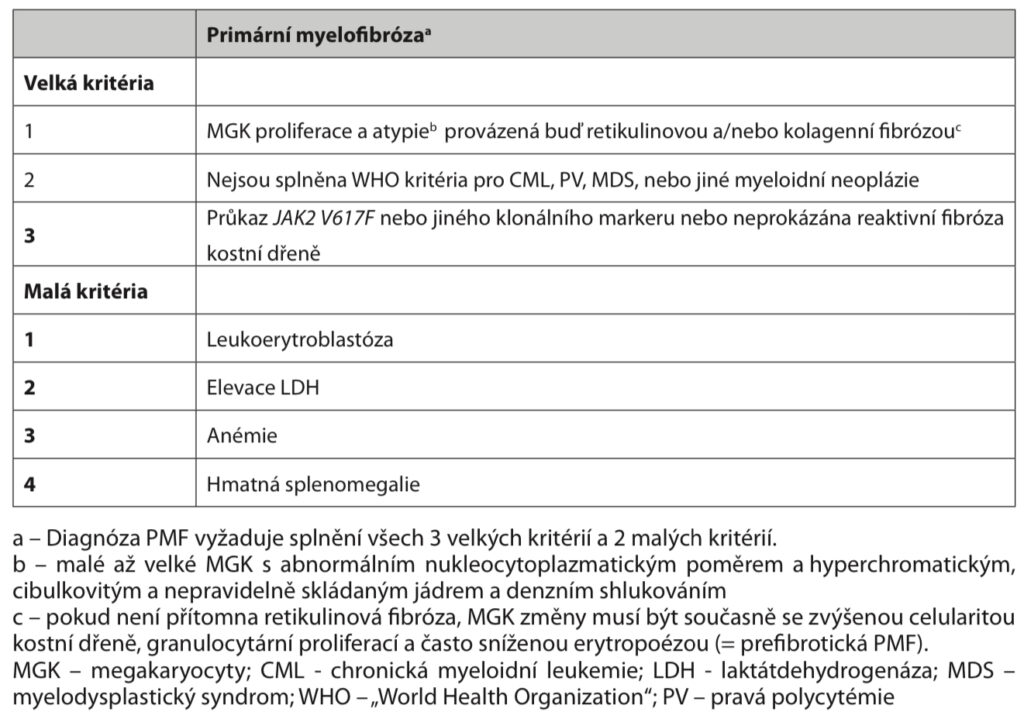
Terapie – v současnosti je jedinou kurativní metodou alogenní SCT, která je indikována u mladších nemocných v pokročilé fázi choroby, ostatní postupy jsou pouze paliativní. V léčbě anémie lze použít erytropoetin, lze zkusit i androgeny, thalidomid, lenalidomid a kortikoidy. Při trombocytóze je indikován anagrelid. Při polyglobulii nebo trombocytóze lze použít erytrocyt – nebo trombocytaferézu. Použití hydroxyurey, interferonu-α i ozáření sleziny je v současnosti na ústupu a je nahrazováno inhibitory JAK2 (ruxolitinib).