APS -I
Definice – autoimunitní polyglandulární syndrom (APS) I typu je vzácné onemocnění (popsáno dosud pouze cca 500 případů). Hlavními komponentami syndromu jsou chronická mukokutánní kandidóza + hypoparathyreoidismus + Addisonova choroba).
Etiologie – AR dědičná mutace AIRE genu (autoimmune regulatory gene, 21. chromozóm). Dosud bylo popsáno > 100 mutací, některé častěji vyskytují v určitých etnických skupinách (Iránští Židé, obyvatelé Sardínie, Finska, Norska a Irska), obě pohlaví jsou zastoupena stejně.
Klinický obraz – typický APS-1 se objevuje velmi časně, typicky v dětství nebo dospívání:
- chronická mukokutánní kandidóza – většinou se manifestuje jako první a nejčastěji postihuje ústa a nehty a vede k postupné atrofii postižených míst. Příčinou je produkce protilátek proti interleukinům (anti-IL17 a anti-IL22), které mají vztah k Th17 populaci T-lymfocytů. Autoprotilátky vedou k poklesu jejich syntézy.
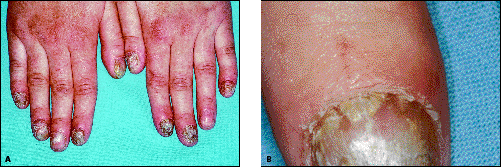
- hypoparatyreoidismus – většinou vzniká po kandidóze (u > 85 % pacientů).
- Addisonova choroba – vzniká až jako poslední (u téměř 80 % pacientů).
- k neendokrinním příznakům lze zařadit alopecie, vitiligo, střevní malabsorpce, perniciózní anémie, chronická aktivní hepatitida a dystrofie nehtů.
Diagnostika – diagnóza je většinou stanovena klinicky v přítomnosti dvou ze tří hlavních komponent. K definitivnímu potvrzení je vhodné genetické vyšetření a stanovení typu mutace AIRE genu.
Terapie – spočívá v terapii jednotlivých složek syndromu.
APS-II
Definice – častější než APS-I s prevalencí 1:50-100 tisíci. Postižení žen je 3x tak časté jako mužů. Projevuje se často až v dospělosti, nejčastěji ve věku 20 – 60 let. Hlavními komponentami syndromu jsou Addisonova choroba (postiženo cca 60 % pacientů) + autoimunitní tyreopatie (Gravesova choroba nebo Hashimotova tyreoiditida, v 15 – 69 %) + diabetes mellitus I. typu (40 – 50 %), dále primární hypogonadismus.
Etiologie – dědičnost není Mendelovská ale polygenní, přičemž hlavní rizikový faktor je lokalizovaný na 6. chromozómu do oblasti kódující HLA komplex (největší predispozicí je HLA-DR3 a HLA-DR4).
Klinický obraz – dán přítomností jednotlivých složek syndromu.
Diagnostika – rozvoj vzácnějších endokrinopatií (např. Addisonova choroba) by měl vést ke pravidelnému screeningu ostatních hormonálních poruch, protože u cca 50 % Addisoniků se během života vyvine další autoimunitní choroba.
Terapie – spočívá v terapii jednotlivých složek syndromu.
Ostatní polyendokrinní syndromy
IPEX (Imunitní dysregulace, Polyendokrinopatie, Enteropatie, X-vázaná dědičnost) – vzácná choroba, pro kterou je typická těžká enteropatie, diabetes mellitus I. typu a kožní změny. Mnoho dětí zemře několik dnů po narození, někteří jedinci se dožijí i 12 – 15 let. Na tuto chorobu je potřeba pomyslet při velmi časném vzniku diabetes mellitus I. typu. Přestože je nutné léčit jednotlivé složky syndromu, zásadním a jediným kurativním postupem zůstává imunosuprese a následná transplantace krvetvorných buněk.
Nádory thymu – thymomy jsou spojeny s několika autoimunitními chorobami, nejčastěji myasthenia gravis (44 %) a aplázie erytrocytů. Většina nádorů je maligních a po jejich resekci dochází k dočasnému ústupu endokrinopatie.
Protilátky proti receptoru pro inzulín – velmi vzácná choroba, přítomnost protilátek proti inzulínovému receptoru působí těžkou inzulínovou rezistenci, dalším znakem je acanthosis nigricans. U 1/3 pacientů se vyskytuje další autoimunitní onemocnění, např. systémový lupus erythematosus nebo Sjögrenův syndrom (tedy i pozitivita ANA, elevace FW, hyperglobulinémie, leukopenie, hypokomplementémie). Glykémie může být vysoká (díky inzulínové rezistenci) i nízká (autoprotilátky mohou i částečně aktivovat inzulínový receptor). Léčba spočívá v potlačení B-lymfocytární odpovědi (rituximab, cyklofosfamid, glukokortikoidy).
Hiratův syndrom (inzulínový autoimunitní syndrom) – je spojen s Gravesovou chorobou a léčbou methimazolem, spolu s určitým HLA haplotypem (nejčastěji HLA-DRB1*04:06). Pacienti trpí závažnými hypoglykemiemi a mají často velmi vysoký titr protilátek proti inzulínu. Základem terapie je vynechání methimazolu.
POEMS syndrom (Polyneuropatie + Organomegalie + Endokrinopatie + M-protein + kožní změny – Skin changes) – U pacientů se nejčastěji projevuje progresivní senzomotorická polyneuropatie, diabetes mellitus (50 %), primární gonadální selhání (70 %), plazmocelulární dyskrázie se sklerotickými kostními lézemi. Nemoc se projevuje ve věku cca 50 let a pacienti obvykle přežívají méně než tři roky. Lze u nich prokázat vysoká hladina VEGF a ostatních zánětlivých cytokinů (IL-1β, IL-6 a TNF-α). Terapeuticky se podává lenalidomid, který vede k poklesu VEGF, nutná je terapie ostatních složek syndromu.