Syntéza hemu – syntéza hemu probíhá v 8 krocích (začátkem je konverze sukcinyl-CoA a glycinu) a je kódována 9 geny (ALA syntáza – ALAS1 a ALAS2). Krok 1, 7, 8, 9 probíhá v mitochondriích, ostatní v cytoplazmě. 85 % vzniklého hemu je spotřebován na hemoglobin, zbytek na CYP, myoglobin a ostatní. Hlavním momentem regulace je inhibice ALAS (1 v játrech, 2 v kostní dřeni) volným hemem.

Klasifikace porfyrií – podle místa poruchy lze porfyrie rozdělit na hepatální a erytropoetické:
- čtyři z pěti hepatálních porfyrií (AIP, HCP, VP a ADP) se projevují akutními neurologickými atakami během dospělosti a akumulací ALA a/nebo PBG (označovány jako akutní). ADP se manifestuje v dětství a dospívaní.
- pátá hepatální porfyrie (PCT) se projevuje kožními puchýři s jizvením (možno i u HCP a VP).
- u erytropoetických porfyrií (CEP, EPP a XLP) se porfyriny akumulují v kostní dřeni a erytrocytech s následnou kožní fotosenzitivitou.
Diagnostika porfyrií – při podezření na porfyrii je indikován test první linie, při jeho abnormálním výsledku se pokračuje specifičtějšími metodami.
Akutní porfyrie – na akutní porfyrie by se mělo pomyslet při vzniku neuroviscerálních příznaků po pubertě (nespecifické bolesti břicha). Prvním krokem je stanovení ALA, PBG a kreatininu v moči ze stejného vzorku (ALA je senzitivnější i specifičtější). Stačí jednorázový vzorek moči, protože 24 hodinový sběr je zbytečný a oddaluje diagnózu. CAVE k nespecifickému zvýšení porfyrinů v moči dochází i při hepatopatiích,
Kožní porfyrie – puchýře spojené s kožními porfyriemi jsou spojeny se zvýšením plazmatických porfyrinů (CAVE někdy zvýšené i při end-stage nefropatii). Tímto lze detekovat EPP a XLP, senzitivnější je stanovení hladiny erytrocytárního protoporfyrinu, nikoliv ale specifičtější. Při pozitivitě je nutné podrobnější došetření.
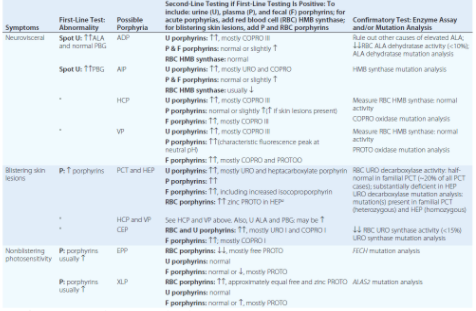
Diagnostika akutní a kožní porfyrie
X-vázaná sideroblastická anemie (XLSA)
Vzniká díky nedostatku ALAS2 (ALA syntázy 2, X chromozóm – postižení zejména muži). Klinická závažnost závisí na reziduální aktivitě ALAS2. Projevuje se refrakterní mikrocytární a hypochromní anémií s anizocytózou během dětství, následuje sekundární hypersplenismus a přetížení železem. Nalézáme hypercelulární kostní dřeň s megaloblastovou přestavbou, prekurzory i porfyriny v moči a séru jsou v normě. Aktivita ALAS2 je snížená. Definitivně lze chorobu potvrdit genetickým vyšetřením ALAS2 genu. Vyzkoušet lze suplementaci pyridoxinem (kofaktor ALAS2). Při rezistenci krevní transfúze terapie a chelatační terapie k prevenci přetížení železem.
Porfyrie při deficitu ALA dehydratázy (ADP) – AR dědičné onemocnění popsané pouze u 20 pacientů, které vzniká díky nedostatku ALAD (ALA dehydratázy). Projevy závisí na reziduální funkci enzymu (symptomatické při < 10 %), připomínají AIP. Všichni pacienti měli zvýšené ALA a koproporfyrin III v moči. V rámci diferenciální diagnostiky je nutné vyloučit hereditární tyrozinémii a intoxikaci olovem (olovo je inhibitorem ALAD). Definitivní potvrzení dá genetické vyšetření. Léčba je podobá AIP.
Akutní intermitentní porfyrie (AIP)
AD dědičné onemocnění (identifikováno > 390 mutací HMBS), které vzniká při poklesu aktivity HMBS (hydroxymetylbilan syntázy) < 50 % s prevalencí 1:20000 (ve Skandinávii 1:10000). Choroba zůstává za normálních okolností latentní do puberty. Jsou známé precipitující faktory vzniku ataky:
- zvýšení syntézy hemu (porfyrinogenní léky, alkohol, nízkokalorická dieta) vede k akumulaci ALA a PBG a vzniku ataky (zakázané léky http://porphyriadrugs.com/)
- zvýšení hladiny progesteronu (období před menstruací, některá kontraceptiva), těhotenství bývá překvapivě dobře tolerováno
Klinický obraz – k podezření na tuto chorobu by měl vést neuroviscerální syndrom vzniklý po pubertě. Ataka může zmizet během několika hodin. Příznaky jsou velmi necharakteristické:
- Bolesti břicha bývají stálé, obtížně lokalizovatelné, mohou mít formu křečí a komplikovat se vznikem ileu, typická je nauzea, zvracení, zácpa/průjem, dysurie nebo retence. Mají neurogenní, nikoliv zánětlivou etiologii (horečka, défense, leukocytóza obvykle chybí). Díky sympatické aktivaci s vegetativním doprovodem vzniká hypertenze, tachykardie, neklid, tremor a nadměrné pocení.
- Neurologické postižení – postižení nervů je méně nápadné než abdominální symptomatologie a projevuje se degenerací axonů (spíše než demyelinizací) s primárním postižením motorických neuronů, senzitivní jsou postiženy méně. Začíná slabostí proximálních svalových skupin, zejména paží a ramen.
- Psychiatrické příznaky (úzkost, nespavost, deprese, dezorientace, halucinace a paranoia) se objevují během akutních atak. Křeče mohou být způsobeny jako neuropatií, tak hyponatrémií (jejich terapie je obtížná, protože většina léků působí exacerbaci AIP, zvážit lze clonazepam).
- Hyponatrémie vzniká postižením hypotalamu (SIADH), průjmem, zvracením nebo nadměrnými ztrátami ledvinami.
Diagnostika – hladina ALA a PBG je zvýšená v plazmě a moči a klesá po několika dnech po podání heminu. Akutní ataku lze diagnostikovat klinicky. Na rozdíl od HCP a VP jsou fekální porfyriny v normě. U asymptomatických heterozygotů je močová exkrece ALA a PBG normální (enzymový defekt lze prokázat jen genetickým vyšetřením).
Častěji je přítomna hypertenze, častější je postižení ledvin i jater, je i vyšší riziko hepatocelulárního karcinomu (nejméně 1 x ročně je indikován UZV jater a hladina AFP).
Terapie – je nutné se vyvarovat vyvolávajícím faktorům, v premenstruální periodě lze podat profylakticky hemin nebo analoga GnRH, která zruší ovulaci a tím i syntézu progesteronu žlutým tělískem. Během akutní ataky je možné podat analgetika ke zvládnutí bolesti, fenotiaziny a malé dávky benzodiazepinů k léčbě nauzey, zvracení, úzkosti a neklidu. U nejlehčích atak (bez hyponatrémie, paréz…) i.v. glukóza (nejméně 300 g denně), u všech ostatních forem i.v. hemin (3 – 4 mg/kg/den). Při brzkém podání je úprava velmi rychlá, u pozdních forem může trvat měsíce až roky.
Jako definitivní řešení je vhodná transplantace jater, zkoušena je i genetická terapie.
Kongenitální erytropoetická porfyrie (CEP – Güntherova choroba) – AR dědičná choroba, dosud popsáno pouze 200 případů. Je způsobena deficitem UROS (uroporfyrinogen syntázy). Těžká fotosenzitivita (vznik puchýřů se vznikem hypo- nebo hyperpigmentovaných křehkých jizev) začíná již v dětství. V kostech a zubech jsou depozita porfyrinů, což působí hnědé zabarvení zubů a jejich fluorescenci v UV záření. Vzhledem k vysokému obsahu porfyrinů v erytrocytech je běžná hemolýza s následnou splenomegalií. Dochází k akumulaci uroporfyrinogenů a koproporfyrinogenů (nejčastěji I) v kostní dřeni, erytrocytech, plazmě, moči a stolici (nejčastěji koproporfyrinogen I). Definitivní potvrzením diagnózy je genetické vyšetření UROS genu. Základem jsou režimová opatření (ochrana před Sluncem a před poraněním kůže), určitý efekt může mít suplementace beta karotenem. V těžkých případech jsou nutné transfúze s následným přetížením železem. Při těžké hemolýze bývá efektivní splenektomie. V některých případech byla účinná transplantace kostní dřeně.

Porphyria cutanea tarda (PCT)
Epidemiologie – nejčastější v regionech s častými infekcemi HCV a HIV.
Patofyziologie – příčinou je absolutní deficit jaterní UROD (uroporfyrinogen dekarboxylázy), choroba se manifestuje při poklesu aktivity pod 20 % normálu. Lze rozeznat řadu vyvolávajících faktorů, které mohou působit synergicky:
- Hepatitida C, HIV
- Nadbytek alkoholu, přetížení železem (mutace C282T a H63D je u PCT častější).
- Substituce estrogeny u žen
- Toxické látky (epidemie PCT v 50. letech v Turecku po kontaminaci pšenice hexachlorbenzenem, dále dioxin).
Klasifikace
- Typ 1 (80 % pacientů) je sporadický (bez přítomnosti mutace).
- Typ 2 (20 % pacientů) je familiární. Genetický defekt postihuje UROD (121 různých mutací), pacienti jsou většinou heterozygoti a jejich aktivita UROD bývá okolo 50 % (samo o sobě nepostačuje k manifestaci). Jsou tedy potřeba ještě další vyvolávající faktory. Penetrance familiární formy je nízká, proto často nelze dohledat pozitivní rodinnou anamnézu.
Klinický obraz – hlavním projevem je vznik puchýřů (nejčastěji na dorzu rukou, ale i jinde), které praskají, vzniká krusta a vše končí jizvou s atrofickým okrskem kůže. Běžná je hypertrichóza a hyperpigmentace zejména obličeje (kosmetický problém). Někdy se kůže po ozáření Sluncem naopak zhrubne a připomíná systémovou sklerodermii.

Diagnostika – v moči může být ALA lehce zvýšené, PBG je v normě. Výrazně zvýšené jsou porfyriny v moči, stolici a plazmě. Typ 2 lze odlišit od typu 1 průkazem snížené aktivity UROD v erytrocytech. Vhodné jsou pravidelné ultrazvukové vyšetření jater a test na AFP k časné detekci hepatocelulárního karcinomu (u PCT častější).
Terapie – nutná režimová opatření (vyvarovat se alkoholu, estrogenům a slunečnímu světlu). Přetížení železem lze odstranit flebotomiemi á 1 – 2 týdny (hladina feritinu by měla být v normálním rozmezí), po dosažení remise nebývají flebotomie již potřeba. K její monitoraci je vhodný odběr plazmatických porfyrinů á 6 – 12 měsíců. Při kontraindikaci flebotomií je možnou alternativou chlorochin a hydroxychlorochin (tvoří komplexy s porfyriny a usnadňují jejich exkreci).
Hereditární koproporfyrie (HCP)
AD dědičná choroba vznikající při poklesu aktivity COPROO (koproporfyrinogen oxidázy, 64 mutací genu CPOX) pod 50 %. Projevuje se většinou po pubertě, častěji u žen a méně závažně než AIP. Podobně jako AIP přichází v akutních atakách (má i stejné vyvolávající faktory), a nezávisle na nich se mohou vyskytovat i kožní léze. Ve stolici a moči je v případě atak výrazně zvýšena koproporfyrinu III (a zejména ve stolici perzistuje i v období remise), méně i ALA a PBG. Neurologické příznaky se léčí jako v případě AIP, léčba kožních lézí flebotomiemi a chlorochinem není efektivní.
Porphyria variegata (VP)
Častá zejména v JAR (v populaci bílých obyvatel 1:350). AD dědičná porucha aktivity PROTOO (protoporfyrinogen oxidázy, 174 mutací genu PPOX). Příznaky: pouze kožní léze podobné PCT (59 %), pouze akutní ataky neuroviscerálních obtíží podobné AIP (20 %) nebo kombinace obojího (22 %). Homozygoti mají výraznější obtíže, které jsou přítomny již od dětství. Během akutních atak jsou zvýšené ALA a PBG, dlouhodobě protoporfyrin ve stolici a koproporfyrin III v moči a stolici. VP lze odlišit od ostatních porfyrií vyšetřením fluorescenčního spektra emitovaného porfyriny v plazmě. Neuroviscerální ataky se léčí jako při AIP, kožní projevy lze zmírnit pouze vyvarováním se slunečnímu svitu (ostatní postupy popisované u PCT nejsou účinné).
Erytropoetická protoporfyrie (EPP)
Etiologie – nejčastější porfyrie u dětí a druhá nejčastější u dospělých. Vzniká při vrozeném poklesu aktivity FECH (ferrochelatázy) pod 35 %.
Patofyziologie – hlavním zdrojem protoporfyrinu jsou retikulocyty kostní dřeně. Po průchodu do krevního oběhu je protoporfyrin většinou volný (myšleno bez komplexu se zinkem) a vázaný na hemoglobin a albumin, přechází krví do jater a je vylučován žlučí a stolicí. Při průchodu cévami kůže působí fotosenzitivitu, ovšem bez vzniku puchýřů. Neindukuje vznik hemolýzy, ale u 5 % pacientů může působit závažné jaterní poškození, protože protoporfyriny nejsou rozpustné a mohou působit cholestázu.
Klinický obraz – projevy fotosenzitivity se liší od ostatních porfyrií. Začíná většinou již v dětství a projevuje se (již několik minut po expozici slunečnímu světlu) bolestivým zarudnutím, svěděním a otokem a může připomínat angioedém. Puchýře nejsou běžné. Při recidivách vznikají chronické změny postižené kůže (lichenifikace, změny pigmentu, hirsuitismus) a nehtů. Mimo sluneční svit neexistují jiné vyvolávající faktory.
Diagnostika – průkaz zvýšeného volného protoporfyrinu zejména v erytrocytech, ale i v kostní dřeni, plazmě, žluči a stolici.
CAVE Ale, ke zvýšení koncentrace erytrocytárního protoporfyrinu dochází i u:
- Otravy olovem
- Deficitu železa
- Hemolýzy
- Všech homozygotích forem ostatních porfyrií a u jejich akutních atak.
- Při nálezu zvýšené hladiny protoporfyrinů + přítomnosti klinických příznaků indikováno potvrzení diagnózy testem rozlišení volného porfyrinu od porfyrinu v komplexu se zinkem.
- Další nálezy – erytrocyty rudě fluoreskují při λ = 620 nm. Porfyriny a jejich prekurzory v moči jsou v normě. Aktivity FECH lymfoblastových kulturách je snížená.
- Definitivní potvrzení dá průkaz mutace při genetickém vyšetření.
Terapie
- Základem je důsledná ochrana před slunečním svitem. U některých osob může toleranci vůči Slunci zvýšit substituce beta-karotenu. Používá se i afamelanotid (analog α-MSH).
- Léčba jaterních komplikací je obtížná. Cholestyramin a aktivované uhlí vážou protoporfyriny do komplexu a měli by přerušit enterohepatální cyklus protoporfyrinů. Z krátkodobého hlediska bývá účinná transplantace jater, choroba ovšem ve štěpu často recidivuje. Někdy mohou pomoci i i.v. hemin a plasmaferéza. Úspěch měla i transplantace kostní dřeně.
- Splenektomie může být prospěšná při výrazné hemolýze a hypersplenismu.