Cystická fibróza
Deficit α1 – antitrypsinu
CAVE Kouření výrazně urychluje progresi choroby.
Diagnostika
- Fyzikální vyšetření – hyperinflace (hypersonorní poklep, oslabené alveolární dýchání).
- Funkční vyšetření plic – obstrukční ventilační porucha, snížena difuzní kapacita CO.
- Sérová koncentrace α1 – antitrypsinu < 0,5 g/l u PiZZ (norma 1,5 – 3,5 g/l), dále průkaz desmozinu v moči (marker degradace elastinu).
CAVE α1 – antitrypsin je protein akutní fáze (zvýšen u zánětu a při zvýšené syntéze IL-1, IL-6 a TNF-α).
- Zobrazovací metody
RTG S+P – známky plicní hyperinflace při emfyzému.

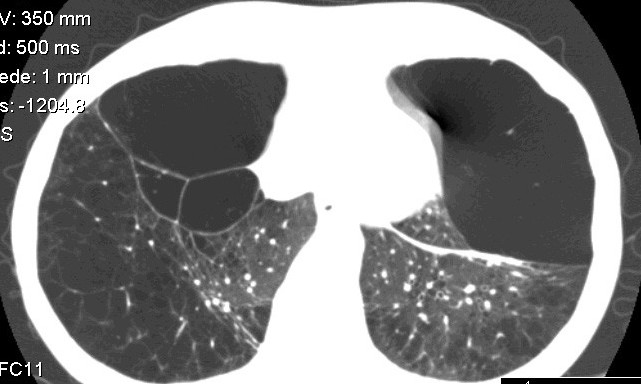
HRCT – bulózní formace, někdy i bronchiektázie
- Jaterní biopsie s průkazem inkluzí v hepatocytech.

Terapie
- Režimová opatření – zákaz kouření, dechová rehabilitace.
- Farmakoterapie – substituce koncentrátu α1 – antitrypsinu, β2 sympatomimetika.
- DDOT při splnění kritérií.
- Tx plic v terminálních stádiích.
Primární ciliární dyskineze
Primární ciliární dyskineze (PCD) je heterogenní choroba s prevalencí odhadem 1:15000 s převážně AR dědičností, známo je > 20 kandidátních genů, ve 38 % jde defekt DNAI1 (9p) a DNAH5 (5p). Příznaky souvisí s chybějící samočistící schopnosti epitelu dýchacích cest (chybění pohybu nebo asynchronní pohyb cílií), která umožňuje zvýšený průnik infekčních agens.
První podezření musí vzbudit nález situs inversus na prenatálním ultrazvuku (v tomto případě je nutné PCD vyloučit vždy). Příznaky:
- Plicní – recidivující záněty horních i dolních cest dýchacích se vznikem bronchiektázií.
- Fertilita – sterilita mužů (porucha pohyblivosti bičíků), nižší fertilita žen (porucha pohyblivosti řasinek vejcovodů).
- Smysly – horší sluch a čich (recidivující záněty, hromadění sekretu).
Kartagenerův syndrom = sinusitis + bronchiektázie + situs viscerum inversus (45 % pacientů s ciliární dyskinezí).
Na PCD je nutné pomyslet v případě sourozence s PCD, situs inversus, respirační tísně novorozenců narozených v termínu, recidivujících infekcí horních i dolních dýchacích cest a středního ucha.
Diagnostika – k diagnóze je potřeba průkaz poruchy funkce řasinkového epitelu pomocí vysokorychlostní videomikroskopie (HSVM) vyšetřuje pohyb řasinek v reálném čase. Vzorek se odebírá kartáčkem z nosní sliznice.
U většiny pacientů s PCD lze pomocí elektronové mikroskopie prokázat i defekty ve struktuře vzorku odebraného z bronchiální sliznice. CAVE Neplatí, že normální elektronmikroskopický nález znamená, že pacient PCD nemá. Genetické testování má zatím pouze omezený význam.
Terapie a prognóza – onemocnění nelze vyléčit. Zásadní je správně prováděná fyzioterapie (dechová rehabilitace, polohová drenáž, poklepová masáž hrudníku) a včasná terapie infekcí (antibiotika, β2 mimetika).
Prognóza je příznivější než u cystické fibrózy, pacienti se dožívají dospělosti, ale mohou u nich vznikat ireverzibilní bronchiektázie a atelektázy.
Tuberózní skleróza
Tuberózní skleróza (TS) je AD dědičná nebo sporadická choroba (u 2/3 de-novo) způsobená mutacemi v jednom ze dvou tumor-supresorových genů (TSC-1, TSC-2), které se podílí na regulaci buněčného růstu a proliferace. Incidence TS je 1 : 5800 živě narozených. Onemocnění postihuje řadu systémů:
1. Kůže – prakticky u 100 % nemocných ve vyšším věku. Kožní léze mají různou podobu (odbarvené kožní skvrny, angiofibromy a fibrózní plaky na čele).

2. CNS – benigní nádory CNS s následnou epilepsii (až 80 % postižených), polovina postižených trpí mentální retardací a poruchou osobnosti (v dětství časté ADHD).

3. Ledviny – u 80 % pacientů, nejčastěji benigní angiomyolipom, vzácněji epiteliální cysty.

4. Další orgány – intrakardiální rhabdomyomy, lymfangiomyomatóza plic.
Diagnostický algoritmus – kritéria vypracována na International Tuberous Sclerosis Complex Conference (2012):

Možná diagnóza: 1 hlavní nebo nejméně 2 vedlejší kritéria.
Obecně je nutná důsledně odebraná osobní a rodinná anamnéza se zaměřením na výskyt příznaků, které mohou souviset s TSC, dále podrobné celotělové vyšetření kůže (doporučeno je vyšetření Woodovou lampou), v případě epilepsie EEG a provedení MRI, event. CT hlavy, ultrazvuk ledvin, v novorozeneckém věku echokardiografické vyšetření a EKG, vyšetření očního pozadí a zvážit CT plic u dospělých žen (plicní lymfangioleiomyomatóza). V případě klinického podezření indikováno genetického vyšetření.
Kauzální léčba není možná. Základem je symptomatická terapie, do medikace zavedeny i m-TOR inhibitory (sirolimus a everolimus). Péče o tyto pacienty vyžaduje multidisciplinární tým. Průběh a závažnost onemocnění je velmi variabilní, uvádí se, že je celkově zkrácena průměrná délka dožití.
TS pěkně zpracovaná také zde: https://www.prolekare.cz/casopisy/cesko-slovenska-dermatologie/2016-2/tuberozni-skleroza-58714