(1/2021)
Definice – chronická lymfatická leukémie (CLL) je monoklonální proliferace zralých B lymfocytů, definovaná jako jejich absolutní množství v periferní krvi > 5 · 109/l (pokud není tento požadavek splněn, stav se označuje jako monoklonální B-lymfocytóza – vyskytuje se 100x častěji než CLL a je považována za prekancerózu, která má 1 – 2 % roční riziko progrese do CLL). Diagnostická kritéria:
- B-lymfocyty v periferní krvi > 5 · 109/l.
- morfologicky ≤55 % atypických buněk (např. prolymfocyty) v periferní krvi.
- typický imunofenotyp (CD5, CD19, CD23, slabě CD20).
Epidemiologie – většinou postiženi starší lidé (medián 71 let) s incidencí 1:18 tisíc, nejčastěji u bílé rasy a 2x častěji u mužů než u žen. Je 8,5x vyšší riziko vzniku choroby při postižení příbuzného prvního stupně.

Etiopatogeneze – nádorovými buňkami jsou zralé B lymfocyty, ale tyto závěry jsou poslední dobou zpochybněny a zvažuje se jestli prekurzorem není multipotentní kmenová buňka nebo paměťové lymfocyty.
Příčina není známá, nicméně nebyla prokázána souvislost s expozicí ionizujícímu záření (na rozdíl od ostatních typů leukémií). Zdá se, že ke vzniku CLL napomáhá celá škála aditivně působících faktorů, např. podíl B buněčného receptoru (BCR), dlouhodobá antigenní stimulace, vliv deregulace miRNA, vliv mikroprostředí lymfatické tkáně apod. Zdá se, že monoklonální B buňky deregulují T lymfocyty s možností vzniku hlubokého imunodeficitu. CLL lze rozdělit dle vzniku na dva základní typy:
- podílí se antigen prezentující buňka (APC), B-lymfocyty prošly zárodečným centrem, kde nastala somatická mutace IgVH (těchto forem je 60 %, vyskytují se rovnoměrně u obou pohlaví a mají lepší prognózu).
- setkání antigenu bez přítomnosti APC a nedochází k somatické mutaci IgVH (těchto forem je 40 %, častěji u mužů, mají špatnou prognózu).
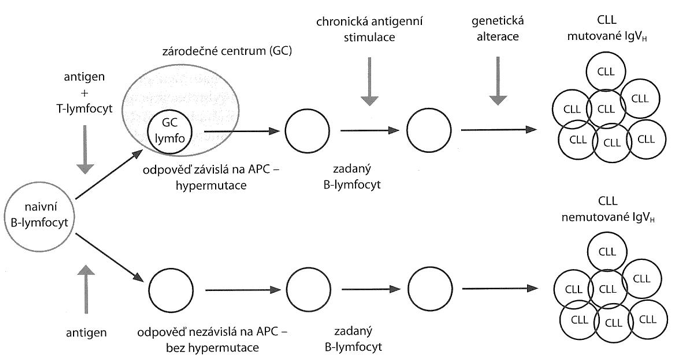
Schéma vzniku CLL.
Často se vyskytují autoimunitní komplikace ve formě hemolytické anémie nebo trombocytopenie. Protilátky jsou polyklonální a jsou tvořeny reziduálními B-lymfocyty. Funkční nedostatečnost patologických B-lymfocytů má za následek defekt protilátkové imunity s větší tendencí k infekcím.
Klinický obraz – u části nemocných je diagnóza zjištěna náhodně, u některých při došetřování příčiny zvětšených lymfatických uzlin. CLL se manifestuje:
- nespecifickými příznaky – malátnost, subfebrilie, noční poty, úbytek hmotnosti.
- generalizovaná lymfadenopatie (uzliny jsou elastické, volně pohyblivé a nebolestivé), splenomegalie, malá hepatomegalie.
- častá je autoimunitní hemolytická anémie a trombocytopenie s příslušnými projevy (anemický syndrom, krvácení).
Diagnostika – ke stanovení diagnózy CLL je potřeba aby byly splněny dvě podmínky:
- absolutní lymfocytóza > 5 · 109/l po dobu 3 měsíců (u malobuněčného lymfomu – SLL je infiltrace uzlin, ale lymfocyty jsou < 5 · 109/l).
- potvrzení typického klonálního imunofenotypu (CD5, CD19, CD23).
Periferní krevní nátěr – převaha zralých lymfocytů s častými Gumprechtovými stíny (kondenzovaný chromatin z poškozených jader fragilních leukemických lymfocytů). Mohou být přítomny větší buňky s výraznějším jádrem (jejich počet by ale neměl přesáhnout 55 % – v tom případě bychom již uvažovali o prolymfocytární leukémii). Ke stanovení diagnózy není potřeba provést analýzu kostní dřeně.
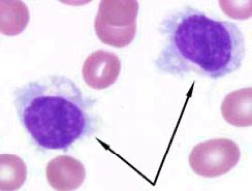
Další doporučená vyšetření:
- průtoková cytometrie ke stanovení imunofenotypu.
- cytogenetické vyšetření pomocí FISH k vyloučení mutací (např. delece 13q je nejčastější, 11q, 17p, trisomie 12).
- stanovení mutačního stavu genů pro IgVH.
- vyšetření mutací genu TP53.
- Coombsův test.
- prognostické testy – beta2-mikroglobulin, thymidinkináza (čím vyšší, tím horší prognóza).
- imunoglobuliny.
- vyšetření kostní dřeně před zahájením léčby.
- k vyloučení lymfadenopatie RTG S+P a sono břicha (u mladších ke zvážení CT).
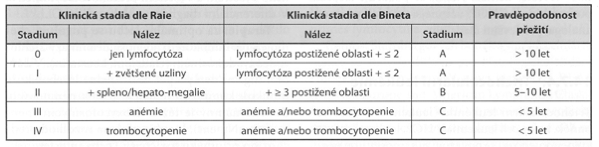
Staging – pokročilost choroby určujeme podle stádií dle Raie/Bineta:
Prognóza – ke stanovení prognózy lze stanovit IPI (mezinárodní prognostický index – dostupné on-line: https://www.mdcalc.com/international-prognostic-index-chronic-lymphocytic-leukemia-cll-ipi. Další faktory:
- stav mutace IgVH (nemutované mají výrazně horší prognózu).
- cytogenetický nález (17p- má horší prognózu s horší odpovědí na léčbu).
- klasifikace dle Raie-Bineta.
Terapie – u počátečních nebo pouze lehčích forem lze vyčkávat se strategií „watch and wait“. Stádium selhávání kostní dřeně (III a IV dle Raie nebo stádium C dle Bineta, tedy anémie nebo trombocytopenie) je indikací k zahájení léčby:
- U mladších nemocných indikujeme režim FCR (fludarabin, cyklofosfamid, rituximab), popř. ibrutinib.
- Při 17p- nebo TP53 popř. IgVH mutací ibrutinib.
- U starších nemocných BR (bendamustin, rituximab) nebo obinutuzumab + chlorambucil.
- Při selhání léčby ke zvážení. transplantace krvetvorných buněk.
Průběh a prognóza – někdy choroba rychle progreduje, jindy zůstává stacionární. Tedy doba přežití 1 -20 let.
Prolymfocytární leukémie
Definice – morfologicky je prolymfocytární leukémie (PLL) charakterizována přítomností > 55 % prolymfocytů v periferní krvi (velké buňky s velkým jádrem).
Epidemiologie – velmi vzácná (incidence cca 1 : 2 milióny).
Klasifikace – dle imunofenotypu lze rozdělit na:
- B-PLL (80 %) – CD20+, CD22+. Věkový průměr 69 let, obvykle se projevuje masivní splenomegalií a B příznaky. Není postižena kůže. Refrakterní k terapii (většinou se podává FCR). Medián přežití 3 roky.
- T-PLL (20 %) – CD2+, CD3+, CD7+. CD4+ a/nebo CD8+. Věkový průměr 61 let. Běžná je masivní splenomegalie, může být hepatomegalie a zvětšení lymfatických uzlin. U 20 % pacientů jsou přítomny kožní příznaky (uzlíky, makulopapulosní rash, vzácně erythrodermie) a dále otoky. Laboratorně je běžná obrovská leukocytóza (> 100 – 200 · 109/l), u 80 % jsou cytogeneticky prokazatelné chromozomální aberace. Lékem volby je alemtuzumab. Medián přežití je 1 – 2 roky.
Detailní informace o diagnostice a léčbě zde: https://www.hematology.cz/doporuceni/klinika-files/Doporuceni_CHS_CLS_JEP-Cervena_kniha.pdf