Sarkoidóza
Definice (ČPFS 2019) – systémové onemocnění neznámé etiologie, pro které je typická přítomnost granulomatózního zánětu v postižených tkáních. Nejčastěji se projevuje bilaterální hilovou lymfadenopatií a/nebo plicními infiltráty. Z mimoplicních postižení je nejběžnější postižení očí a kůže, lymfatických uzlin, motorického ústrojí a jater, vzácněji sleziny, slinné žlázy, srdce, nervového systému a jiných orgánů. Akutní formou je Löfgrenův syndrom.
Epidemiologie – častěji v chladných pásech (nejčastěji Skandinávie – 3x častěji než v ČR, v USA více černá než bílá rasa), méně v tropech a tam, kde je vysoký výskyt TBC, muži: ženy 1 : 2, ve věku 30 – 50 let (muži mladší), více nekuřáci, v ČR je prevalence 1:1400.
Etiologie – systémové granulomatózní onemocnění neznámé etiologie, pravděpodobně imunitní reakcí na neznámý antigen u geneticky predisponovaných jedinců – uvažuje se o celé řadě infekcí (mykobakterie, borelie, Propionibacterium acnes a další) nebo hypersenzitivní reakci na některé kovy (berylium, nikl, hliník), talek apod. Vzniká oligoklonální buněčná odpověď s akumulací CD4+ Th1 lymfocytů, které aktivují makrofágy, zásadní je i účast Th17 lymfocytů.
Patogeneze – mechanismus není přesně znám. V aktivní fázi tvorby granulomu dominují T lymfocyty a monocyty s makrofágy, v chronické převažují fibroblasty. Dochází ke vzniku granulomů se vznikem mnohojaderných buněk vznikajících z aktivovaných makrofágů s vystupňovanými známkami sekreční činnosti. V centru granulomu dominují CD4+ Th-lymfocyty, na periferii cytotoxické CD8+ Tc-lymfocyty a fibroblasty. Granulomy dále produkují celou řadu substancí. např. 1,25-dihydroxycholekalciferol (hyperkalcémie a hyperkalciurie), ACE, neopterin, solubilní receptor pro IL-2.
Typickým nálezem je zvýšení poměru CD4+/CD8+ v BAL a následně opačně snížení tohoto poměru v periferní krvi. Díky tomu je utlumena pozdní tuberkulinová reakce prokazatelná kožními testy. Ústřední cytokin je TNFα.
Klinický obraz – nemoc je často asymptomatická a je náhodným RTG nálezem. Klinicky manifestní sarkoidóza se projevuje:
- syndromem systémové zánětlivé odpovědi (subfebrilie, horečka, únava, malátnost, hubnutí, alterace celkového stavu)
- respiračními příznaky (dráždivý chronický kašel, dušnost, výjimečně bolest na hrudi)
Akutní sarkoidóza (Löfgrenův syndrom)
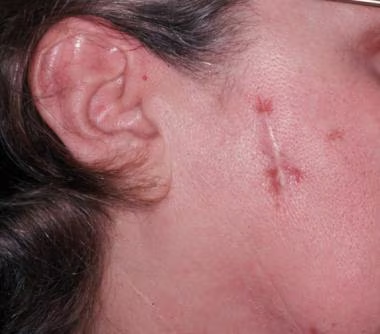
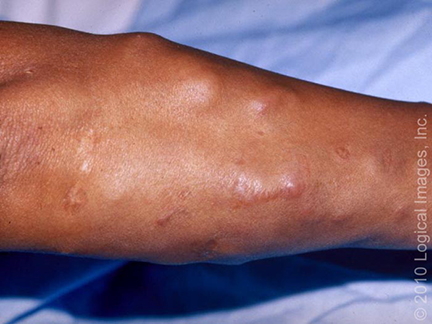
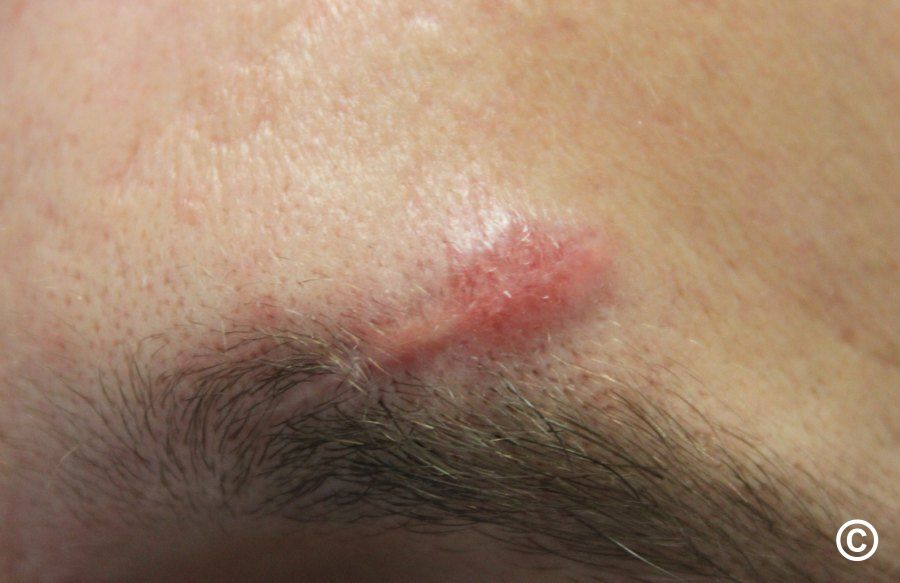
= horečka + faryngitida + artralgie (nejčastěji hlezna, postižený kloub je bolestivý, teplý a výrazně oteklý) + perimaleolární otoky + erythema nodosum (bérce, kotníky) + hilová lymfadenopatie + negativní tuberkulinová reakce. Dále sarkoid v jizvě (zčervenání a zduření starých jizev).
K zapamatování: chřipka při které bolí klouby, hlavně hlezna a jsou oteklé a s erytémem.
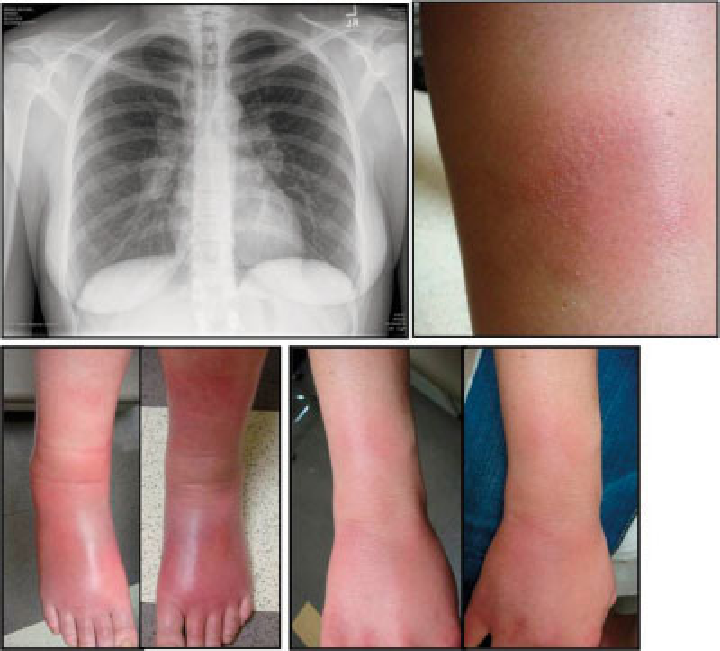
Chronická sarkoidóza
Chronická sarkoidóza trvá alespoň 2 roky. Lokalizace postižení je následující:
- 1. Plíce (90 % postižených) – dušnost, neproduktivní kašel, bolesti na hrudi s intersticiálním plicním procesem až fibrózou plic. Bývají postiženy i dýchací cesty projevující se dušností, suchostí sliznice, chrapotem a bolestmi na hrudi.
- 2. Játra (50 – 80 % postižených) – hepatomegalie nebývá častá, většinou je jen mírná elevace jaterních enzymů.
- 3. Oči (25 – 50 % postižených) – fotofobie, slzení, bolest, porucha vizu až slepota, uveitis, skleritis, konjunktivitis.
- 4. Klouby, svaly (30 % postižených) – artralgie, artritida, myopatie.
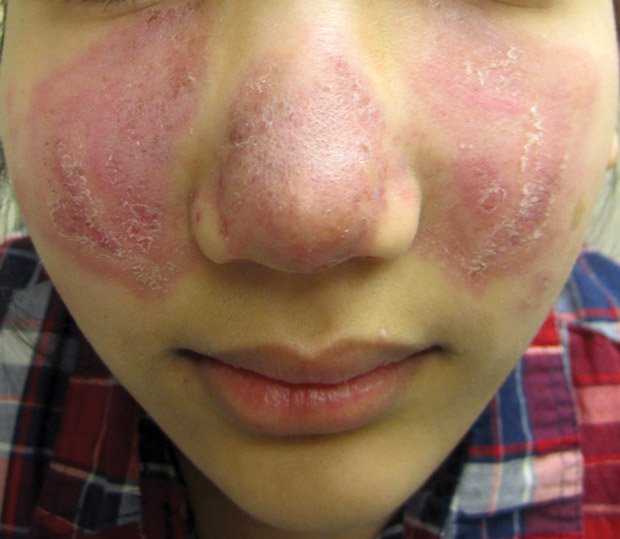
- 5. Kůže (25 % postižených) – erythema nodosum, lupus pernio, podkožní uzly, sarkoid v jizvě.
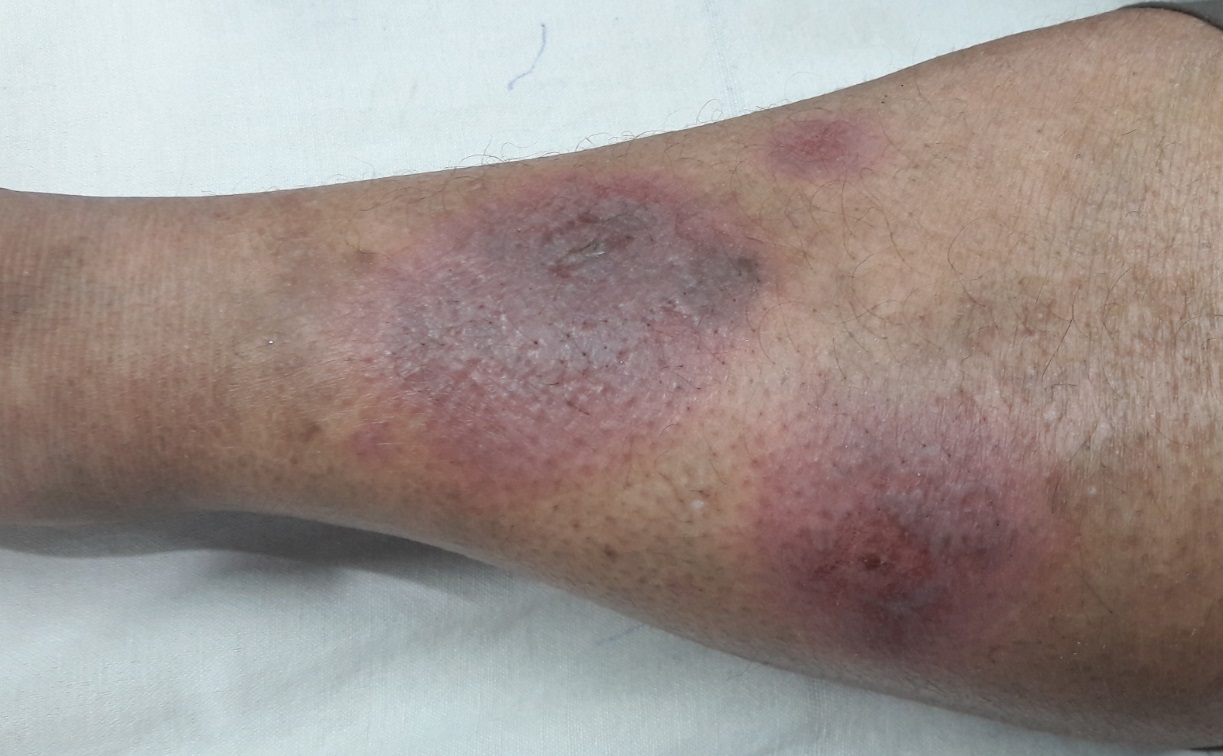
- 6. Kosti – kostní cysty vyplněné granulomy (nejčastěji střední a distální falangy) projevující se bolestí.
- 7. Nervový systém (5 – 10 % postižených) – paréza facialis, bolesti hlavy, křeče, meningitis, encefalitis
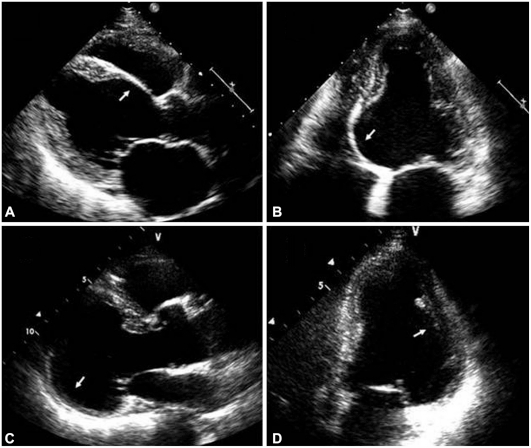
- 8. Srdce (5 % postižených, ale až ve 20 % autopsií) – projevuje se zejména převodními poruchami , komorovými arytmiemi a srdečním selháním. U mladých jedinců s pokročilou AV blokádou je vždy třeba vyloučit sarkoidózu. EKG i echokardiografie mohou být normální, zásadní je MRI srdce, kde je nález LGE v neischemické lokalizaci (chybí subendokardiálně a je nepravidelně rozmístěn, nejvíce v bazálních segmentech levé komory) a v akutní fázi lze prokázat edém myokardu. Dalším důležitým vyšetřením je PET CT (méně PET-MRI) s důležitou dietní přípravou (vyloučení sacharidů a hodně tuků k přesmyknutí fyziologického glukózového mechanismu). K definitivnímu průkazu choroby je potřeba histologická verifikace pomocí EMB, popřípadě u pacientů s extrakardiální manifestací typická klinická manifestace podpořená nálezem na MRI nebo PET. Metodou léčby jsou kortikoidy, při neúčinnosti jako léčba druhé linie azathioprin, mykofenolát mofetil , cyklofosfamin, při neúčinnosti pak antagonisté TNF (infliximab, adalimumab). U pacientů se synkopou, setrvalou komorovou arytmií nebo sekundárně je indikována implantace ICD.
- 9. Lymfatický systém – zvětšení uzlin, splenomegalie.
- 10. Endokrinní systém (velmi vzácně) – hyperkalcémie, hyperkalciurie, diabetes inspidus.
- 11. GIT (velmi vzácně) – dysfagie, bolesti břicha, ikterus.
- 12. Příušní slinné žlázy neboli febris uveoparotidea (vzácné) – teplota + přední uveitida + zduření slinné žlázy + obrna tváře = Heerfordtův syndrom.
- 14. Ledviny – sekundárně postiženy při dlouhodobé hyperkalcémii (nefrokalcinóza, nefrolitiáza, selhání ledvin).
CAVE Ne každá choroba s nálezem epiteloidních granulomů je sarkoidóza!!!
Diagnostika
1. Laboratorní metody – nespecifické zvýšení FW, CRP, anémie chronických chorob, leukopenie, lymfopenie. Dále je přítomna hyperkalcémie a hyperkalciurie (tvorba vitamínu D3, granulomy). Existující i určité specifické markery sarkoidózy – sACE, sIL-2R, neopterin (marker aktivity interferonu gama).
2. Imunologické testy
- imunoregulační index: CD4+/CD8+ T lymfocyty v krvi snížen
- tuberkulinový test je negativní v 70 %, pozitivita diagnózu nevylučuje
- Kveimův test – extrakt ze sleziny pacienta nemocného sarkoidózou se aplikuje subkutánně. Pozitivita při objevení granulomů za 4 – 6 týdnů. Při současné aplikaci kortikoidů může být falešná negativita. V současnosti se neprovádí.
3. Zobrazovací metody
a) RTG S+P a CT hrudníku – 5 stádií (viz níže)
- stádium 0 – bez patologických změn
- stádium 1 – symetrická hilová lymfadenopatie bez postižení plic
- stádium 2 – zvětšení hilových uzlin s postižením plic. V průběhu choroby nastává zmenšení hilových uzlin s progresí plicního postižení („útěk do plic“) což je patognomonické.
- stádium 3 – pouze plicní postižení bez zvětšených uzlin
- stádium 4 – plicní fibróza

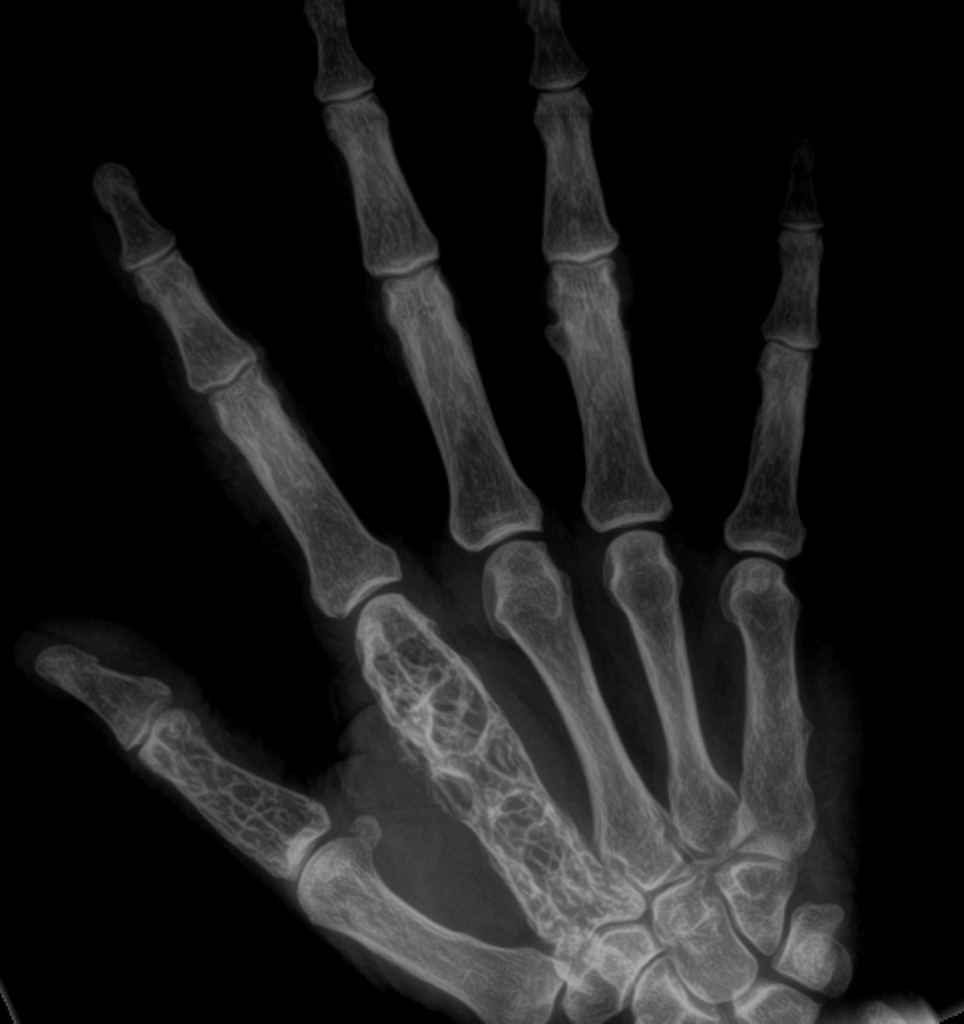
b) RTG rukou – periartikulární osteoporóza, mnohočetné cystoidní projasnění (ostitis multiplex cystoides Jüngling).
c) Ostatní
- bronchoskopie – překrvení, někdy nažloutlé uzlíky, při BAL nález vysokého počtu lymfocytů (CD4+/CD8+ T lymfocyty v BAL typicky zvýšený)
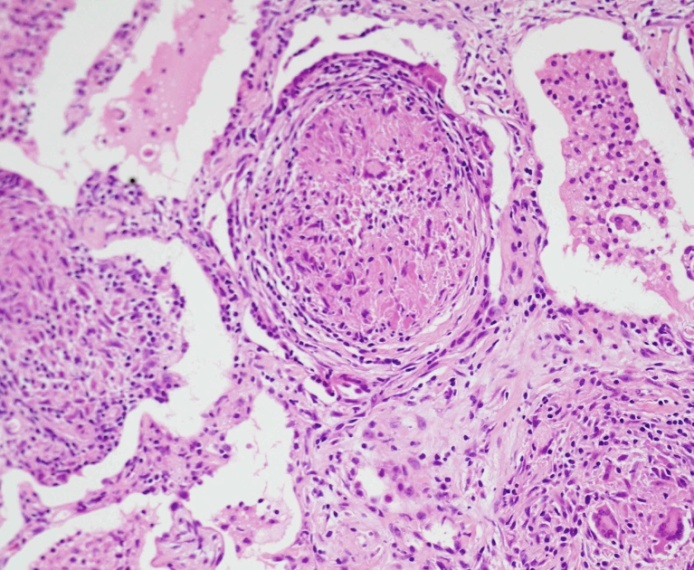
- histologické vyšetření -vhdoné k podpoření diagnózy. Při transbronchiální biopsii, bronchiální punkci nitrohrudních uzlin, VATS, mediastinoskopii, při mimoplicní lokalizaci biopsie z postiženého orgánu. Nález nekaseifikujících epiteloidních granulomů.
- funkční vyšetření plic – v časných stádiích diskrepance mezi RTG nálezem a funkčním vyšetřením, na počátku porucha difuze a krevních plynů při zátěži.
- dále zobrazovací vyšetření dle postiženého orgánu.
Terapie
- Watch and wait – někdy léčbu není nutné zahajovat. V praxi je léčeno cca polovina nemocných. Ke spontánní remisi dochází u 55 – 90 % pacientů stadia I, u 40 – 70 % ve stadiu II a u 10 – 20 % ve stadiu III. Nejvíce remisí je pozorováno během prvních šesti měsíců.
- Léčba I. linie (kortikosteroidy) – signál k zahájení léčby systémovými kortikoidy je symptomatické plicní onemocnění nebo těžká ventilační porucha, hyperkalciurie a orgánové poškození (CNS, srdce, kůže nereagující na topickou terapii…). Lokální kortikoidy jsou indikovány u kožní formy a erythema nodosum, dále při postižení očí a dýchacích cest. Inhalační kortikoidy lze použít pouze při izolovaném postižení plic po tříměsíčním předléčení systémovými kortikoidy. Použít lze prednison iniciálně 20 – 40 mg/den s vyhodnocením léčby po 3 – 6 měsících.
- Léčba II. linie – při nemožnosti udržet remisi dávkou 10 mg prednisonu denně se přidávají další imunosupresiva (metotrexát, hydroxychlorochin, azathioprin, leflunomid, cyklofosfamid, mykofenolát mofetil).
- Léčba III. linie (biologická léčba) – při selhání nebo intoleranci léků II. linie (anti TNFα – infliximab, adalimumab).
- Ostatní léčba – při splnění indikací dlouhodobá domácí oxygenoterapie, při postižení mozku radioterapie, při postižení srdce implantace ICD, dále rehabilitace.
- Transplantace se provádí i u sarkoidózy jater, ledvin a srdce.
Prognóza – prognóza je většinou příznivá. Dominuje postižení plic a nitrohrudních uzlin, mimoplicní postižení bývá zjištěno u 50 % nemocných. Ze skupiny pacientů s klinickou manifestací dochází u > 50 % ke spontánní remisi, 30 – 50 % ke zhojení po aplikaci kortikoterapie, 20 – 30 % případů má chronický průběh
Blíže viz http://www.pneumologie.cz/guidelines/. Aktualizace 2019.
Ostatní granulomatózy
1. Inhalace organického prachu se vznikem hypersenzitivní pneumonitidy
2. Silikóza
Etiologie – chronické fibronodulární onemocnění způsobené protrahovanou (10 – 20 let) inhalací prachu obsahujícího oxid křemičitý (SiO2). Postižení jsou horníci rudných nebo kamenouhelných dolů, pracovníci v lomech, kameníci, tuneláři, skláři.
Patofyziologie – SiO2 působí při fagocytóze na alveolární makrofágy a polymorfonukleáry, které uvolňují volné radikály, chemotaktické a růstové faktory což vede k proliferaci fibroblastů, následné plicní postižení je pak reparováno lokální fibrózou.
Klinický obraz – prostá silikóza je často asymptomatická, mohou být přítomny pouze příznaky chronické bronchitidy. V pozdějších stádiích vzniká námahová dušnost, krepitus nad plicními bázemi a paličkovité prsty.
Diagnostika
- pracovní anamnéza
- RTG S+P – nejdříve mnohočetná ložiska, které později splývají (hlavně střední plicní pole) a přecházejí do plicní fibrózy. Patognomonické jsou skořápkovité kalcifikace v lymfatických uzlinách.


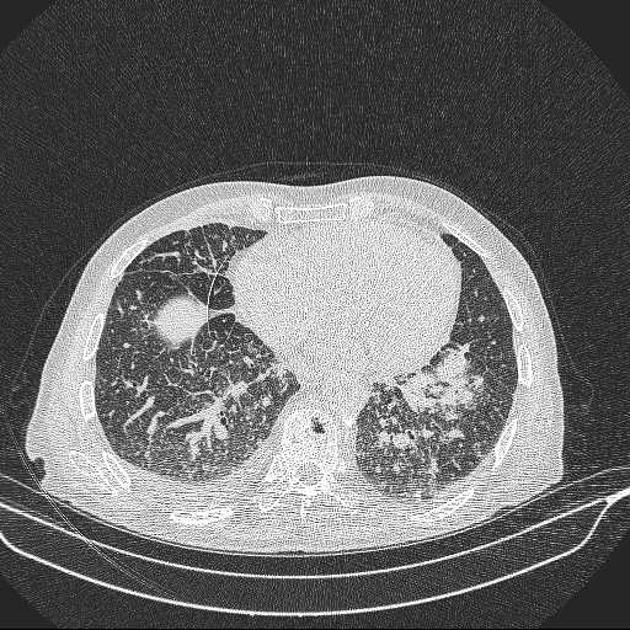
- HRCT plic – malé uzly s opacitami mléčného skla a ztluštělými septy s obrazy mnohoúhelníku („crazy paving„). Nálezy na: https://radiopaedia.org/articles/silicosis
- funkční vyšetření plic – postupně vznik restriktivní poruchy
- plicní biopsie – ve sporných případech
Terapie a prognóza – hlavní je zástava expozice včetně zákazu kouření, dechová rehabilitace, symptomatická terapie (expektorancia, bronchodilatancia, ATB při infekci, kyslík), kortikoidy mohou zpomalit progresi. V indikovaných případech transplantace plic. Choroba progreduje i po ukončení expozice, je zvýšené riziko karcinomu plic a TBC.
3. Granulomatózní vaskulitidy
Granulomatóza s polyangitidou (Wegener)
Definice – ČPFS 2016: Granulomatóza s polyangitidou (GPA) je chronické multisystémové onemocnění nejasné etiologie, které je charakterizováno nekrózami, tvorbou granulomů a vaskulitidou horních a dolních dýchacích cest, nekrotizující glomerulonefritidou a systémovou vaskulitidou postihující malé a méně často i střední tepny.
Epidemiologie – incidence GPA je 1:70 tisíc, prevalence 1:35 tisíc, častěji bílá rasa, lehce častěji postiženi muži, nejčastější věk 40 – 60 let.
Etiopatogeneze – etiologie je neznámá. Pravděpodobně hypersenzitivní reakce na některé mikrobiální agens v terénu genetické predispozice. Úzce souvisí s přítomností protilátek proti cytoplasmě neutrofilů (anti neutrophil cytoplasmic antibodies – ANCA), které se dělí na C-ANCA (cytoplasmatické, častěji proti proteináze-3) a P-ANCA (perinukleární, častěji proti myeloperoxidáze). Vazbou ANCA na cílové struktury dochází k degranulaci neutrofilů, poškození endotelu buněk, poškození malých cév a ke vzniku vaskulitidy. Nález ANCA protilátek v plasmě, a to obvykle podtypu c-ANCA proti PR3, se vyskytuje v iniciální fázi asi u 50 % pacientů, ve fázi generalizace kolem 90 %.

Otázkou je původ ANCA, jednou z hypotéz je vznik protilátek původně namířených proti S. aureus, které jsou i kompatibilní k proteináze -3.
Důležitá je i role TNF-α, která stimuluje neutrofil, který až poté exprimuje proteinázu 3 na svém povrchu a zpřístupní ji tak pro ANCA.
Klinický obraz – klasické trias: postižení horních cest dýchacích + dolních cest dýchacích + ledvin.
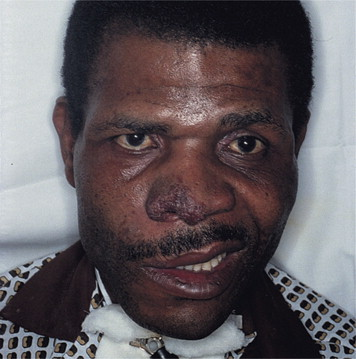
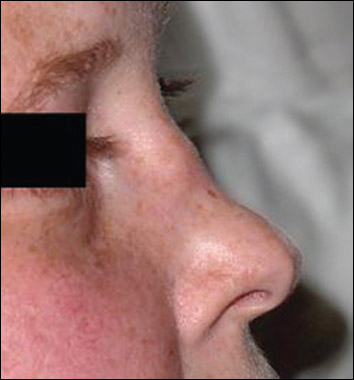
Horní dýchací cesty (90 %) – často výrazná paranazální bolestivost s výtokem purulentního nebo krvavého sekretu s možnou přítomností slizničních ulcerací. Komplikace jsou blokáda Eustachovy trubice (vznik otitis media), perforace nosní přepážky (vznik sedlovitého nosu).
Dolní dýchací cesty (90 %) – často kašel a bolesti na hrudníku. Difuzní alveolární hemoragie může vést k život ohrožující hemoptýze. Mohou být ulcera laryngu s následným vznikem subglotické tracheální stenózy (stridor). Stále častější bývá endobronchiální postižení s nálezem bronchitických fenoménů.
Ledviny (75 %) – lehká glomerulonefritida s proteinurií, hematurií a erytrocytárními válci s rychlou následnou progresí do srpkovité RPGN (nutné co nejdříve zahájit léčbu).
Systémové příznaky (40 %) – febrilie, únava, slabost, malátnost, úbytek hmotnosti.
Oči – obstrukcí ductus lacrimalis nebo očnice granulomy (lehká konjunktivitida, dacryocystitida, episkleritida, skleritida, granulomatózní sklerouveitida, vaskulitida cév ciliárního aparátu až vznik retroorbitálních granulomů, vedoucí k exoftalmu).

Kůže – papuly, vezikuly, palpovatelná purpura, ulcerace nebo podkožní noduly. Biopticky je přítomna vaskulitida, granulomatóza nebo obojí.
Nervový systém – kraniální neuritida, mononeuritis multiplex nebo vzácně mozkovou vaskulitidou a/nebo granulomatózou.
Srdce – perikarditida, koronární vaskulitida nebo vzácně kardiomyopatie.
CAVE Granulomatóza s polyangitidou je trombofilním stavem!
Diagnostika
1. Laboratorní nález – zvýšení FW, mírná anémie, leukocytóza, trombocytóza (reaktant akutní fáze), lehká hypergamaglobulinémie (zejména IgA), mírná elevace RF.
- C-ANCA (proti proteináze-3) – pozitivní u 90 % pacientů s aktivní chorobou, u 60 % v remisi, pozitivita proti myeloperoxidáze jen minimálně.
2. Biopsie a histologické vyšetření – definitivní diagnóza je dána průkazem nekrotizující granulomatózní vaskulitidy u pacientů s odpovídajícími klinickými příznaky. Biopsie horních cest dýchacích s nálezem granulomů a nekrózy (ne vždy přítomna vaskulitida), biopsie plic je nejprůkaznější (téměř vždy nález granulomatózní vaskulitidy), biopsie ledvin s nálezem pauciimunní glomerulonefritidy. Mimo toto typické postižení může být postižen vaskulitidou nebo granulomem (nebo obojím) jakýkoliv orgán.
3. Zobrazovací metody – základním vyšetření je RTG S+P, který by měl být doplněn CT. Nález je pestrý s průkazem četných nodulů, nehomogenních infiltrátů, kavit, někdy s hladinou. Difuzní alveolární krvácení se projevuje neostře ohraničenou kondenzací. Radiologické nálezy zde: https://radiopaedia.org/articles/granulomatosis-with-polyangiitis. K průkazu hypermetabolismu glukózy lze použít PET CT.
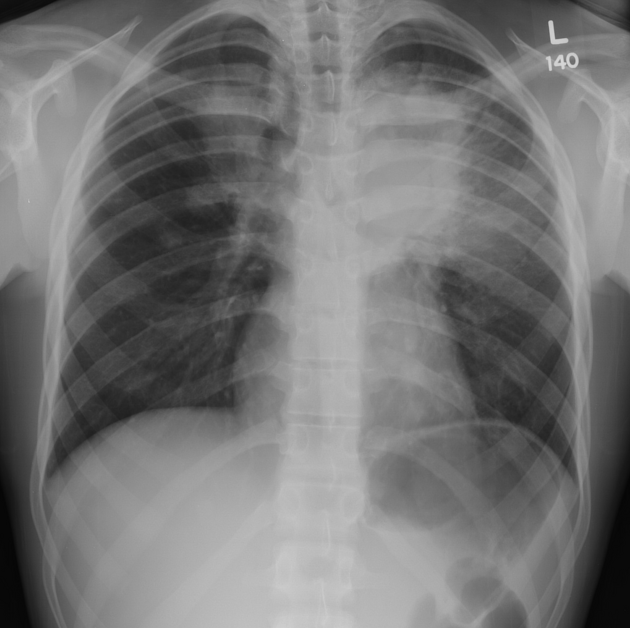
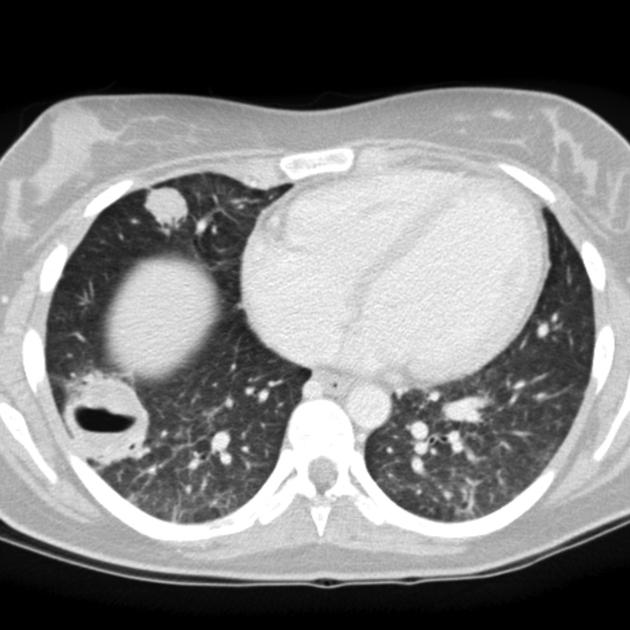
4. Funkční vyšetření plic – při vyšetření difúze může být patrné snížení difuze, naopak u difuzní alveolární hemoragie je hodnota difuze zvýšená. Může být přítomna restrikční poruchu, ale nález může být i v normě. Při postižení dýchacích cest může být obstrukce
5. Bronchoskopie a BAL – při podezření na difuzní alveolární hemoragii je indikována diagnostická BAL (červená lavážní tekutina, event. mikroskopicky nález siderofágů). Často jsou nalézány i pseudotumory z granulační tkáně, které obturují průsvit průdušek.
Diferenciální diagnostika – podobně se může projevovat Goodpastureův syndrom, relabující polychondritida, tumor horních cest dýchacích a plic, infekce (histoplazmóza, mukokutánní leishmanióza, rhinosklerom), neinfekční granulomatóza, angiocentrické imunoproliferativní léze odvozené z T-buněk, lymfomatoidní granulomatóza odvozená z B-buněk, spojená s EBV infekcí, dlouhodobý abusus kokainu
Terapie
1. Indukční (k dosažení remise)
- Prednison – první měsíc 1 mg/kg (max. 60 – 80 mg) se snahou o snížení dávky < 10 mg denně. Při těžkém průběhu indikovány parenterální pulzy methylprednisolonu.
- Cyklofosfamid – parenterálně (s redukcí dávky při CHRI) po dobu 3 – 6 měsíců s kontrolou krevního obrazu á 1 – 2 týdny.
- V současnosti je možné podat rituximab (anti-CD20) již v indukční fázi léčby (zejména při kontraindikaci cyklofosfamidu). Přestože není přítomna toxicita vůči močovému měchýři, je riziko nežádoucích účinků stejné jako u cyklofosfamidu – těžké mukokutánní léze, vzácně progresivní multifokální leukoencefalopatie, reaktivace hepatitidy B (před zahájením terapie je nutné serologické vyloučení hepatitidy). Popř. je možné podat methotrexát.
- Plazmaferéza – indikována u pacientů s RPGN s hladinou kreatininu > 512 μmol/l.
2. Udržovací (k udržení remise) – po 3 – 6 měsících se přeruší léčba cyklofosfamidem a přejde se na udržovací léky prednison + :
- Azathioprin – v dávce 2 mg/kg/den. Při intoleranci:
- Methotrexát – v jedné dávce 0,3 mg/kg týdně (ale ne více než 15 mg týdně) p.o. nebo s.c. Při dobré toleranci po 2 týdnech lze týdně navyšovat dávku o 2,5 mg na konečnou dávku 20 – 25 mg/týden a ponechat na této dávce. Vždy nutná substituce kyselinou listovou den po aplikaci methotrexátu.
Délka podávání udržovací terapie není pevně stanovena a je nutné ji posuzovat individuálně (ale udržovací léčba minimálně 1,5 – 2 roky).
Prognóza – bez léčby je střední doba přežití 6 měsíců. Při léčbě glukokortikoidy a cyklofosfamidem je dosaženo výrazného zlepšení stavu u > 90 % pacientů, remise u 75 % pacientů, 5 letého přežití u 60 % pacientů. U 50 – 70 % pacientů v remisi dochází k jednomu nebo více relapsům choroby.
CAVE K hodnocení aktivity choroby by neměl být používán titr ANCA protilátek, který s ní nekoreluje.
Eosinofilní granulomatóza s polyangitidou
Definice – dříve syndrom Churg – Straussové. Onemocnění charakterizované asthmatem + hypereozinofilií v periferní krvi + tkáňovou eozinofilií + histopatologickým obrazem nekrotizující vaskulitidy a extravaskulárních granulomů. Na rozdíl od polyarteritis nodosa postihuje spíše cévy malého kalibru a souvisí s asthma bronchiale.
Epidemiologie – incidence je velmi nízká (1:200 tisíc – 2 milióny/rok), prevalence 1: 100 tisíc, nejčastěji ve středním věku a lehce častěji muži.
Patogeneze – nejasná, převaha Th2 imunologické odpovědi (určitou roli hraje i Th1 se vznikem granulomů). Přítomno snížení T regulačních lymfocytů produkujících IL-10. Časté protilátky cANCA, které zjištěny u cca poloviny nemocných.
Klinický obraz – několik fází:
- Prodromální – chronická rhinitis s nosními polypy či astmatem, může trvat řadu let.
- Časná systémová fáze – objevují se eozinofilie a prchavé plicní infiltráty.
- Pozdní systémová fáze:
- nespecifické – horečka s váhovým úbytkem
- plicní – masivní infiltráty bez rozpadu, někdy difuzní postižení intersticia
- ledvin – erytrocyturie ev. i malá proteinurie, u malé části pacientů selhání ledvin
- srdeční – myokarditida s rozvojem srdečního selhání
- nervové – periferní mononeuropatie
Mimotechnická pomůcka pro zapamotování mononeuropatie: WARDS PLC (Wegener, Amyloidóza/AIDS, Revmatoidní artritida, Diabetes, Sarkoidóza, Polyarteritis nodosa, Lepra, Carcinom/Churg Strauss).
Diagnostika – kritéria (v současnosti se od těchto kritérií postupně ustupuje a diagnostika se soustředí na klinický obraz). Diagnóza je dána splněním minimálně 4 ze 6 kritérií:
- Astma bronchiale
- Prchavé plicní infiltráty dle RTG S+P
- Postižení vedlejších nosních dutin
- Eozinofilie
- Extravaskulární eosinofilní infiltráty (biopticky verifikované)
- Periferní neuropatie
Laboratorní vyšetření: zvýšení zánětlivých markerů (FW, CRP, leukocyty), anémie chronických chorob, pozitivita cANCA (proti proteináze 3).
Biopsie – zánětlivé změny na středních a malých cévách s eozinofilním infiltrátem.
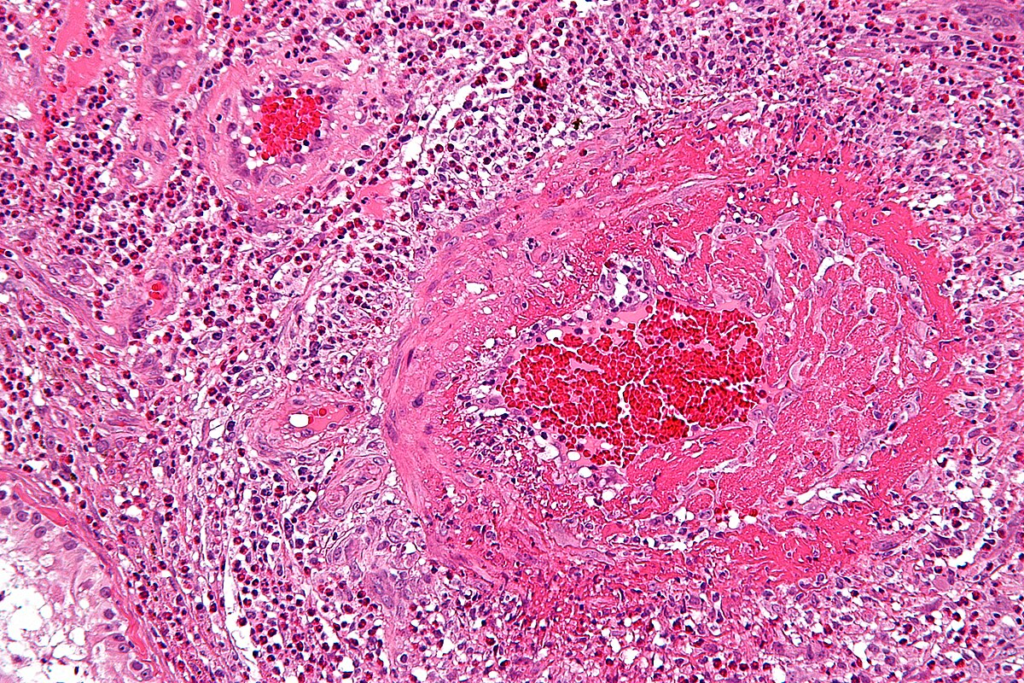
Terapie – léčbou první volby jsou glukokortikoidy (prednison 40 – 60 mg/kg/den s postupnou detrakcí). Léčbou druhé volby u pacientů s těžším průběhem nebo kortikorezistentních cyklofosfamid.
Prognóza – po zavedení kortikoidů se přežití výrazně zlepšilo.
Před zavedením kortikoidů umírala polovina pacientů do 3 měsíců od prvních projevů vaskulitidy a 5 let přežívalo méně než 5 % pacientů. V současnosti pět let přežívá díky kombinované léčbě více než 75 % nemocných. Nejčastější příčinou smrti v akutní fázi onemocnění je infarkt myokardu nebo srdeční selhání. Výskyt pozdních relapsů nemoci je možný, ale není tak častý jako u jiných vaskulitid.